網絡結構及增塑劑對PBT彈性體玻璃化轉變的影響①
2019-03-27 07:49:12苗建波邊利峰朱宏春時志權詹發祿
固體火箭技術 2019年1期
關鍵詞:質量
苗建波,邊利峰,朱宏春,時志權,詹發祿
(1.中國航天科工集團公司六院四十六所,呼和浩特 010010;2.中國航天科技集團公司八院八○六所,湖州 313000)
0 引言
現代導彈武器,尤其是戰術導彈武器,應用環境和作戰平臺復雜多樣,要求固體推進劑在整個使用溫度范圍內具備良好的力學性能,這就要求所使用的粘合劑體系具有較低的玻璃化溫度。PBT推進劑是以3,3-雙(疊氮甲基)氧丁烷-四氫呋喃(BAMO-THF)共聚醚為粘合劑的復合固體推進劑,是繼NEPE推進劑之后的新一代固體推進劑,其兼具有能量高、安全性能好的優點,在先進戰術發動機上有良好的應用前景。但PBT分子鏈中含有大量較大側基,分子鏈柔順性降低,玻璃化溫度較高。因此,PBT推進劑使用溫度下限局限在-45 ℃左右,無法滿足防空反導、空空導彈等戰術發動機-55~70 ℃的寬溫使用要求。
對于降低固體推進劑粘合劑玻璃化溫度,國內外采取的技術途徑主要是網絡結構調控和選用增塑效率高的增塑劑。Manser等[1-3]進行了BAMO與THF、GAP、AMMO和NMMO等共聚物合成,降低了聚合物玻璃化溫度。雙(2,2-二硝基丙醇)縮甲醛/乙醛(BDNPF/A,質量比1∶1)是偕二硝基類含能增塑劑,在疊氮類推進劑有廣泛的應用,代替硝酸酯增塑劑時,可改善固體推進劑低溫力學性能[4]。Provatas[5]研究了多種含能增塑劑對pGLYN粘合劑的增塑效率。結果表明,N-丁基硝氧乙基硝胺(Bu-NENA)增塑效率優于1,2,4-丁三醇三硝酸酯(BTTN)、BDNPF/A。趙昱等[6]開展了Bu-NENA增塑GAP研究,增塑比由0.6增加至1.6,彈性體玻璃化溫度由-59 ℃降至-71 ℃。鄧蕾等[7]采用分子動力學(MD)方法模擬研究了PBT粘合劑與BDNPF/A、Bu-NENA、1,3-二疊氮基-2-乙基-2-硝基丙烷(DAENP)等含能增塑劑的相溶性,與BDNPF/A相比,Bu-NENA、DAENP與PBT相溶性更好,且PBT/Bu-NENA和PBT/DAENP混合體系玻璃化溫度更低。而關于PBT彈性體低溫性能調控的實驗研究,鮮有報道。
本文從網絡結構與增塑兩方面,研究降低PBT彈性體玻璃化溫度的途徑,以期改善PBT推進劑低溫性能。
1 實驗
1.1 原料
PBT,黎明化工研究院生產。固化劑甲苯二異氰酸酯(TDI)、異佛爾酮二異氰酸酯(IPDI)、六亞甲基二異氰酸酯(HDI),交聯劑丙三醇(GY),均為化學純以上。硝酸酯類增塑劑為自制,其他增塑劑為黎明化工研究院生產。
1.2 彈性體的制備
彈性體配方:在考查PBT預聚物結構對彈性體玻璃化溫度影響時,以丙三醇為交聯劑、IPDI為固化劑,保持交聯參數ρ=0.5(式(1))、固化參數R=1.2(式(2))不變,制備彈性體;在考查固化劑對彈性體玻璃化溫度影響時,以BAMO/THF摩爾比為50/50、相對分子質量為5400的PBT預聚物為粘合劑,改變固化劑種類,保持其他組分及R、ρ值不變,制備彈性體;在考查增塑劑對彈性體玻璃化溫度影響時,以BAMO/THF摩爾比為50/50、相對分子質量為5400的PBT預聚物為粘合劑,以丙三醇為交聯劑、IPDI為固化劑,改變增塑劑種類及增塑比,保持其他組分及R、ρ值不變,制備彈性體。
(1)
(2)
彈性體制備過程:將粘合劑、固化劑、交聯劑、增塑劑按上述設計比例混合,在50 ℃下混合均勻,真空脫氣,然后澆入250 mm×150 mm×3 mm模具內,在60 ℃烘箱中恒溫固化5~7 d,取出放入干燥器中待用。
1.3 測試儀器及條件
1.3.1 DSC法測定璃化溫度
預聚物的玻璃化溫度采用德國耐馳204 F1型差示掃描量熱儀(DSC),依據GJB/J 5229—2003測定。
1.3.2 DMA法測定玻璃化溫度
彈性體的玻璃化溫度采用美國TA Q800型動態熱機械分析儀(DMA)測定,正弦力場,頻率1 Hz,升溫速率2 ℃/min。
1.3.3 拉伸性能的測定
采用美國Instron5965型材料試驗機,依據GB/T 528—2009測定彈性體的最大抗拉強度和伸長率。
2 結果與討論
2.1 網絡結構對PBT玻璃化溫度的影響
2.1.1 BAMO/THF比例對PBT玻璃化溫度的影響
PBT是BAMO-THF共聚醚(見圖1)。圖2為實驗獲得的PBT預聚物Tg(DSC)與BAMO質量分數的依賴關系,其中5種PBT預聚物相對分子質量都約為5400。由圖2可知,PBT預聚物玻璃化溫度處于兩種均聚物的玻璃化溫度之間,因為它具有居中的鏈剛性和居中的內聚能密度。PBT預聚物玻璃化溫度與BAMO質量分數存在很好的對應關系,符合Gorden-Taylor方程,即隨著BAMO質量分數的增大,PBT預聚物玻璃化溫度相應升高。
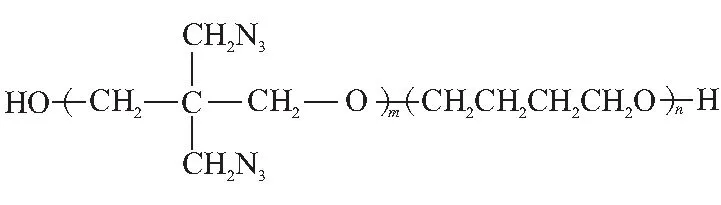
圖1 PBT分子結構式Fig.1 Molecular structural formula of PBT

圖2 PBT預聚物玻璃化溫度與BAMO質量分數關系Fig.2 Tg of PBT prepolymervs mass fraction of BAMO unit
圖3為DMA法測定的BAMO/THF摩爾比分別為30/70、50/50的PBT彈性體(不含增塑劑)玻璃化轉變曲線。在DMA曲線中,通常取損耗模量E"的峰值溫度為玻璃化溫度,記為Tg(DMA)。50/50的PBT彈性體Tg(DMA)為-39.7 ℃,30/70的為-54.1 ℃,BAMO摩爾分數從50%降至30%,PBT彈性體玻璃化溫度降低了14.4 ℃,與PBT預聚物玻璃化溫度變化規律一致。

圖3 PBT30/70和PBT50/50彈性體DMA曲線Fig.3 DMA curves of elastomer made of PBT
2.1.2 數均相對分子質量對PBT玻璃化溫度的影響
表1列出了3種數均分子質量(Mn)不同的PBT預聚物的理化性能,三者的氮含量均在37%左右,表明三者BAMO/THF摩爾比均為50/50。按高聚物自由體積理論[8],每個鏈末端均比鏈中間部分具有較大的自由體積。因此,含有較多鏈末端的試樣比含有較少鏈末端的試樣要更冷,才能達到同樣的自由體積,即具有更低的玻璃化溫度。表1中,PBT預聚物Tg(DSC)隨Mn的變化符合這一規律。
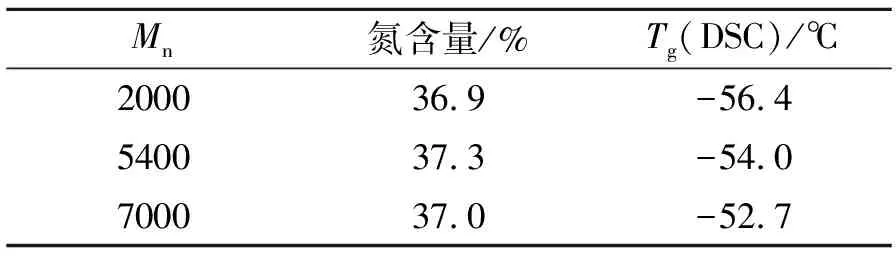
表1 不同數均相對分子質量的PBT預聚物理化性能
圖4為由表1中PBT預聚物制備的彈性體(不含增塑劑)DMA曲線。由圖4可知,PBT彈性體Tg(DMA)隨預聚物Mn增大而降低,Mn由2000增大至5400,PBT彈性體Tg(DMA)降低了16.1 ℃,但繼續增大至7000時,PBT彈性體Tg(DMA)僅降低了1.2 ℃。分析其原因,Mn為2000時,PBT彈性體中交聯點間分子鏈很短,軟鏈段運動能力受到交聯點的極大限制,玻璃化溫度高;隨著PBT預聚物Mn增長至5400,彈性體交聯點間分子鏈變長,軟鏈段運動能力顯著增強,因而彈性體玻璃化溫度顯著降低。當軟鏈段長度超過一定長度時,預聚物Mn對PBT彈性體玻璃化溫度的影響已經很小。

圖4 不同數均相對分子質量的PBT彈性體DMA曲線Fig.4 DMA curves of PBT elastomer with different Mn
2.1.3 固化劑種類對PBT彈性體玻璃化溫度的影響
PBT彈性體中硬段由異氰酸酯與小分子交聯劑組成,其對彈性體玻璃化溫度也有一定影響。采用圖5所示的3種固化劑分別制備彈性體(不含增塑劑),采用DMA測定其玻璃化轉變,結果見圖6。
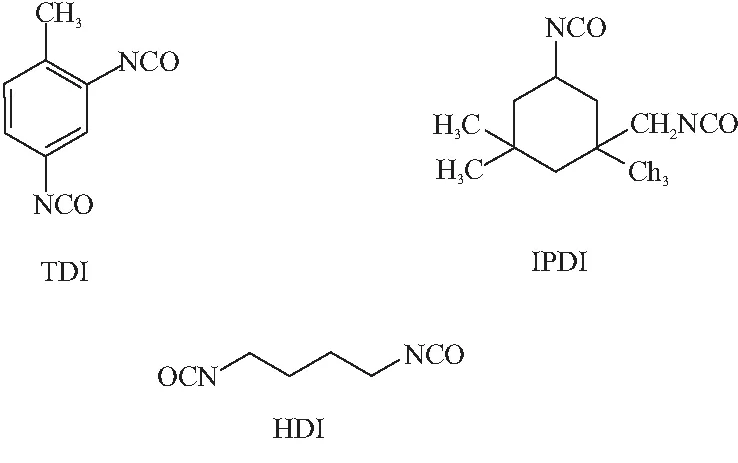
圖5 不同種類異氰酸酯分子結構式Fig.5 Molecular structural formula of different types of isocyanate
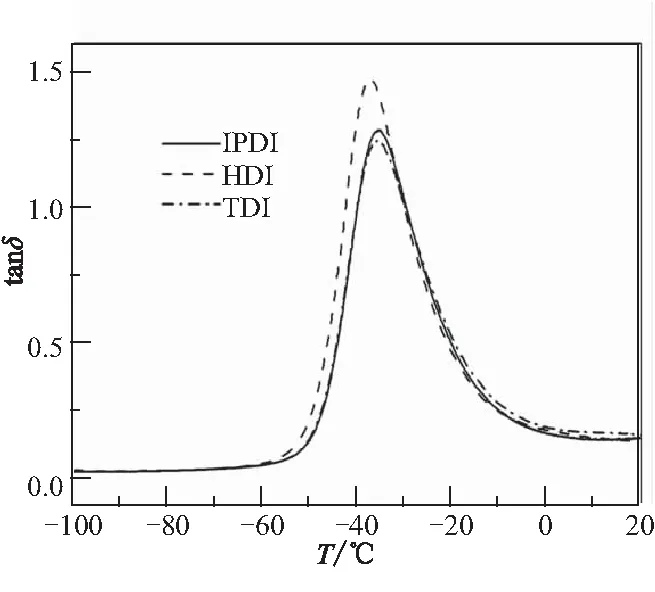
圖6 不同異氰酸酯制備PBT彈性體DMA曲線Fig.6 DMA curves of PBT elastomer containing different isocyanates
在DMA曲線中,tanδ峰高低代表著鏈段運動能力的強弱。在圖6中,3種PBT彈性體玻璃化轉變的tanδ峰高依次為HDI>IPDI>TDI。顯然,這與固化劑分子結構有關。HDI是脂肪族異氰酸酯,具有對稱的柔性鏈結構,HDI形成的硬段對PBT軟段限制作用弱,因而軟段在更低的溫度下發生玻璃化轉變,且tanδ峰高最大;IPDI是環脂族異氰酸酯,結構不對稱,兩個—NCO處在六元環的間位,形成的交聯點不利用于PBT軟段的內旋轉,對軟段運動有很強的限制作用,因而彈性體tanδ峰高明顯低于HDI;TDI是芳香族異氰酸酯,形成的硬段內聚能密度大,且含有20%非對稱2,4-TDI異構體和80%對稱的2,6-TDI,對PBT軟段運動能力限制作用很大。因此,玻璃化溫度在三者中最高,tanδ峰值最低。
關于固化劑分子結構對彈性體玻璃化轉變的影響,文獻[9]已通過分子動力學進行了模擬研究,模擬結果與本文實驗結果一致。
2.2 增塑劑對PBT玻璃化溫度的影響
2.2.1 增塑劑對PBT的增塑效率
表2為PBT預聚物與候選增塑劑的理化性能,其中溶度參數δ采用基團加和法計算得到。從表2看出,NG、BTTN極性較強,與PBT溶度參數差值Δδ約為3 (J/cm3)1/2。TEGDN、TMETN、BDNPF/A、Bu-NENA與PBT溶度參數接近,Δδ≤2 (J/cm3)1/2。惰性增塑劑DOS、LDZ極性較弱,與PBT溶度參數也較接近,Δδ≤2 (J/cm3)1/2。

表2 PBT及增塑劑的理化性能[10,11]
圖7為實驗獲得的PBT預聚物/增塑劑混合體系Tg(DSC)與增塑劑質量分數的關系,圖中實線代表理想線性關系。其中,在質量分數0.30~0.50時,NG、NG/BTTN、TEGDN、TMETN與PBT預聚物的混合體系出現了不同程度的相分離。因此,只給出質量分數較低時的Tg值。
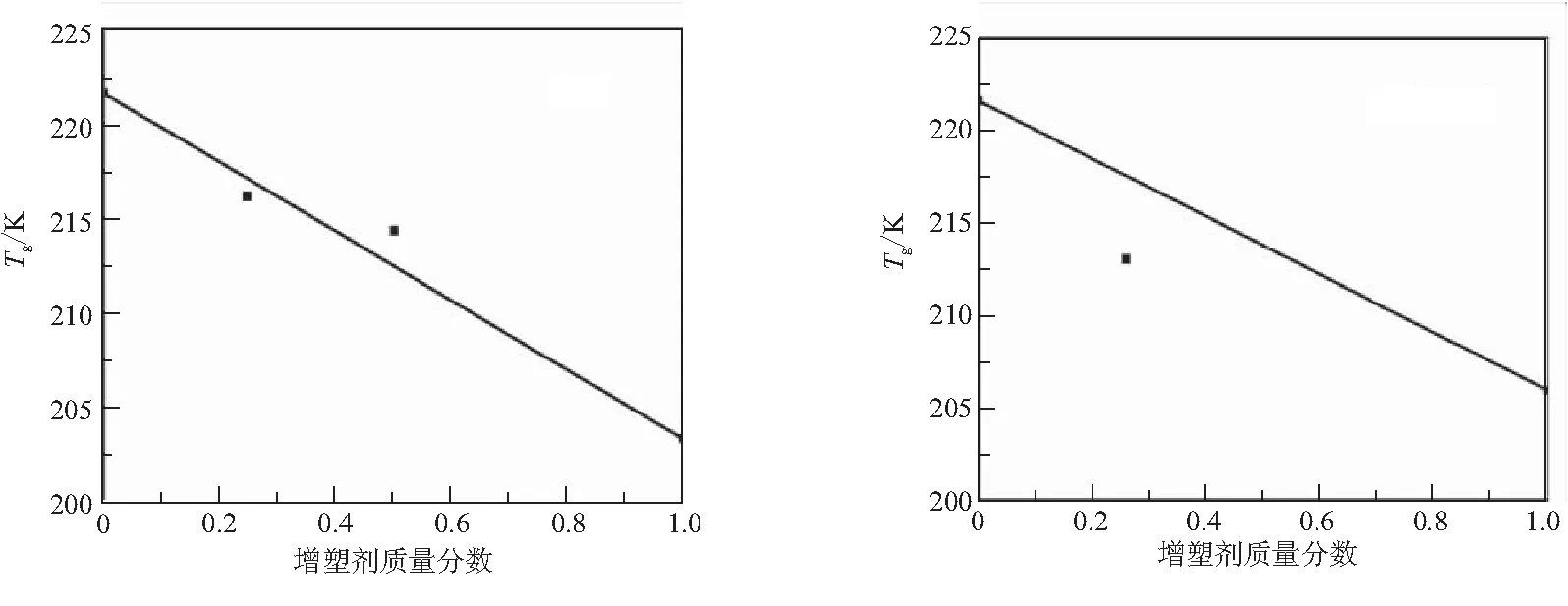
(a)NG (b)NG/BTTN
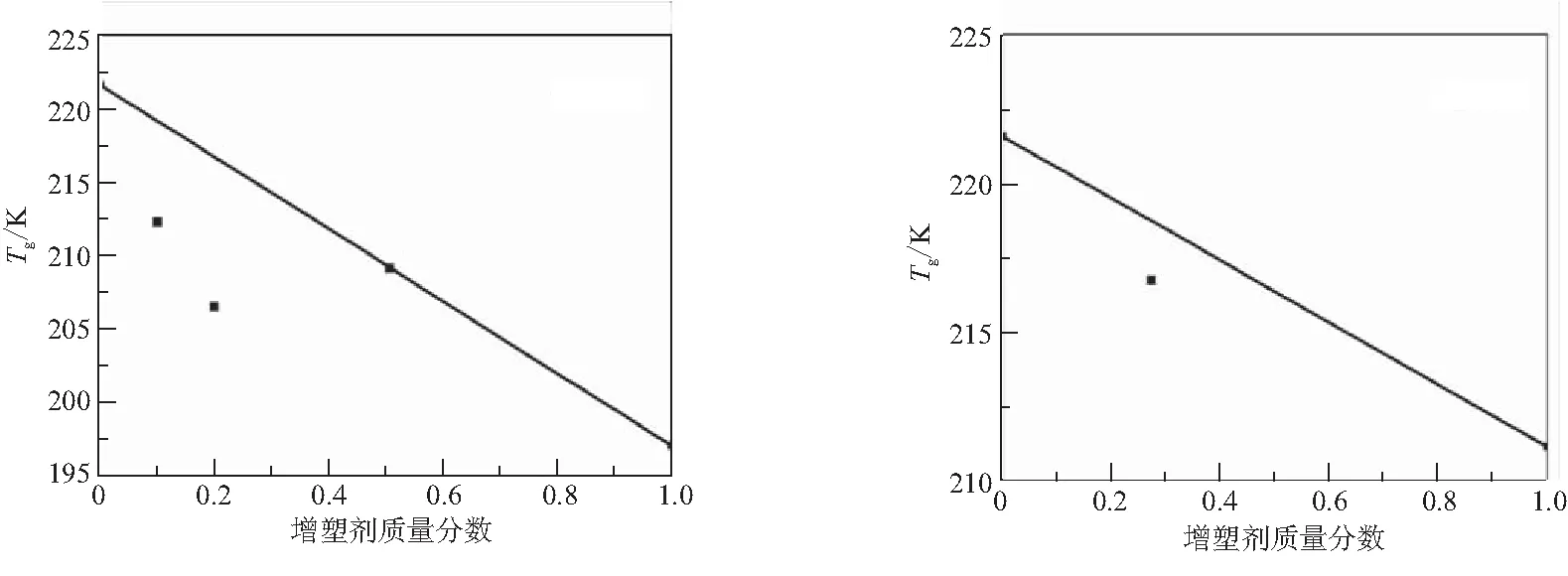
(c)TEGDN (d)TMETN

(e)Bu-NENA (f)BDNPF/A
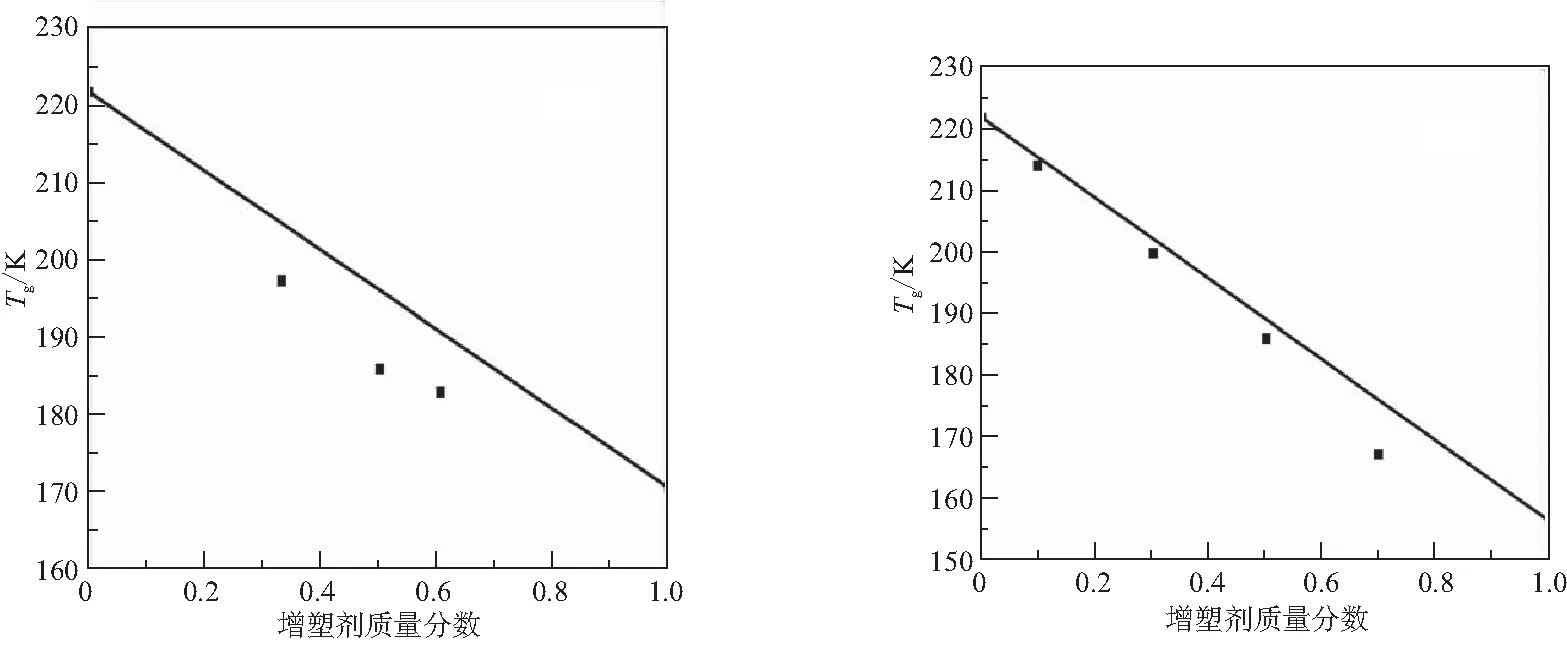
(g)DOS (h)LDZ
圖7表明,不同增塑劑的增塑行為與理想線性關系的偏離情況差異較大。4種硝酸酯增塑劑,質量分數較低時Tg落于理想直線下方,隨著質量分數增加Tg向理想直線靠攏,甚至落在直線上方。雖然TEGDN、TMETN與PBT理論計算的溶度參數接近,但在高質量分數時出現了不相溶。BDNPF/A、LDZ增塑行為基本符合線性關系。Bu-NENA、DOS增塑PBT體系向低溫方向偏離線性關系程度較大。Gordon和Taylor由玻璃態到橡膠態的自由體積變化定義Tg,增塑劑類小分子增加聚合物鏈段間距離,自由體積增加,提高聚合物鏈段運動能力而降低Tg。Tg偏離線性關系與自由體積的改變相關,即增塑劑導致了自由體積增加量大于理想值。
定義增塑效率ε(式(3)),表3列出了上述增塑劑對PBT的增塑效率。硝酸酯類增塑劑在質量分數較高時出現了相分離,因而表3給出了質量分數為0.25的ΔTg;其他增塑劑,給出了質量分數為50%的ΔTg。
ε=ΔTg/wd
(3)
式中 ΔTg為PBT/增塑劑混合體系相對于PBT預聚物的玻璃化溫度降低值;wd為混合體系中增塑劑質量分數。
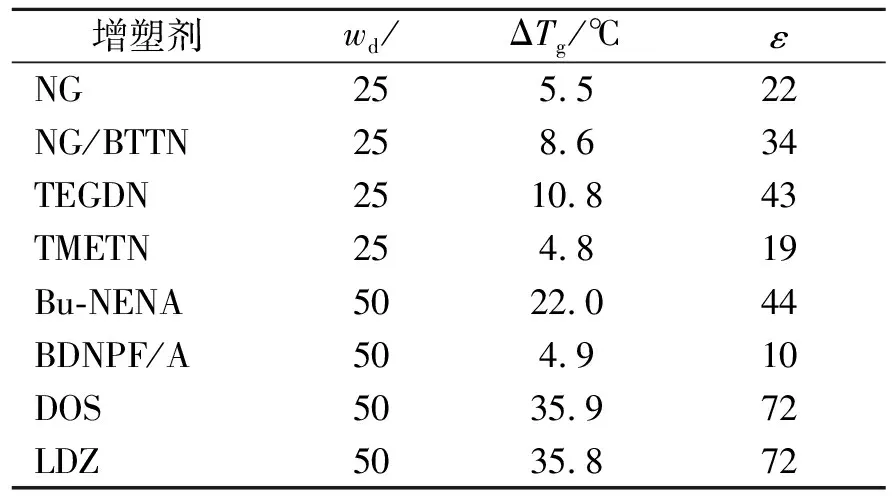
表3 不同增塑劑對PBT的增塑效率
由表3可知,惰性增塑劑DOS、LDZ對PBT的增塑效率遠高于含能增塑劑;在含能增塑劑中,Bu-NENA增塑效率最高。不難看出,增塑劑的增塑效率與其他PBT的相溶性及自身玻璃化溫度的高低有關。根據國內外的研究[12-13],增塑劑的玻璃化溫度高低與其分子結構因素相關,主要包括相對分子質量、柔性基團(柔性側基或脂肪族長鏈骨架)、受阻結構(空間位阻或—ONO2、—N—NO2、—NO2、—CO等極性基團)。增塑劑相對分子質量越小,分子結構中含有較多的柔性基團、較少的受阻結構,增塑劑玻璃化溫度越低。NG、BTTN、TMETN分子中都含有3個—ONO2極性基團,分子主鏈很短,3個—ONO2存在相互限制作用(空間位阻),因此三者都具有較高的玻璃化溫度,且三者與PBT相溶性都較差,僅在質量分數較低時對PBT有一定增塑作用。TEGDN雖然分子兩端為—ONO2極性基團,但是其有較長的脂肪族骨架,因而其玻璃化溫度顯著低于NG、BTTN、TMETN。遺憾的是,雖然TEGDN與PBT的溶度參數相近,但實驗表明,兩者相溶性差,增塑劑質量分數較高時出現了相分離。BDNPF/A與PBT相溶性好,BDNPF/A分子中具有較長的脂肪族長鏈(相對于TMETN、BTTN),但其較大的相對分子質量和4個—NO2極性側基使其玻璃化溫度較高(與TMETN、BTTN相當),限制了其增塑潛能。Bu—NENA雖然分子含有1個—N—NO極性側基和1個—ONO2極性端基,但其分子結構存在兩方面有利因素:一是分子一端中含有柔性丁基,端丁基有較大的自由體積,能提高分子的構象變化與運動能力;二是分子是近似線性結構,—N—NO與—ONO2距離較遠,兩者之間的相互限制作用較弱。這兩方面因素,使得Bu-NENA玻璃化溫度在研究的含能增塑劑中最低,加之Bu-NENA/PBT相溶性好,Bu-NENA增塑效率高于其他5種含能增塑劑。DOS由于分子具有脂肪族長鏈骨架,具有非常低的玻璃化溫度。LDZ非常低的玻璃化溫度主要得益于具有3個柔性端基丁基。DOS、LDZ極性低,與PBT相溶性好,對PBT的增塑效率非常高。
2.2.2 PBT/Bu-NENA彈性體低溫性能
表4列出了PBT彈性體及增塑后的玻璃化溫度和低溫力學性能。

表4 PBT彈性體低溫性能
從表4看出,增塑比為0.5時,Bu-NENA使PBT彈性體Tg(DMA)降低至-52.6 ℃;增塑比為1.5時,Tg(DMA)可進一步降低至-68.3 ℃。在降低PBT彈性體玻璃化溫度方面,Bu-NENA遠優于BDNPF/A,但不足之處在于:Bu-NENA/PBT彈性體抗拉強度顯著低于BDNPF/A /PBT彈性體。
3 結論
(1)PBT預聚物結構單元比例、數均相對分子質量均對PBT彈性體玻璃化轉變有重要影響。預聚物中BAMO摩爾分數由50%降低至30%,PBT彈性體玻璃化溫度降低了約14 ℃。預聚物數均相對分子質量由2000提高至5400,PBT彈性體玻璃化溫度降低了約16 ℃,數均相對分子質量繼續增大,玻璃化溫度降低較小。
(2)固化劑分子結構對PBT彈性體玻璃化轉變有一定影響,具有對稱柔性鏈結構的異氰酸酯制備的PBT彈性體玻璃化溫度略低。對在TDI、IPDI和HDI三種固化劑中,對軟段運動能力限制作用強弱依次為HDI (3)增塑劑對PBT的增塑效率主要取決于其與PBT的相溶性及自身玻璃化溫度的高低。NG、BTTN、TEGDN、TMETN與PBT相溶性差,增塑比較高時,PBT/增塑劑混合體系有明顯的相分離。惰性增塑劑DOS、LDZ對PBT增塑效率非常高,可大幅降低玻璃化溫度。在研究的含能增塑劑中,Bu-NENA對PBT增塑效率最高,在增塑比為1.5時,彈性體玻璃化溫度由-39.7 ℃降低至-68.3 ℃,遠優于現在常用的增塑劑BDNPF/A。
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
