美國藥品主文件年報遞交要求與實踐
2019-05-22 07:31:54施一然
中國藥業 2019年10期
關鍵詞:信息
施一然,梁 毅
(中國藥科大學國際醫藥商學院,江蘇 南京 211198)
從1939年美國食品藥物管理局(FDA)接收第1份美國藥品主文件(DMF),到1989年FDA發布世界首個DMF指南[1],美國DMF制度的規范化用了半個世紀的時間,形成了較成熟的原料藥管理制度[2]。美國DMF制度的創新之處在于:1)原料藥廠商只需向FDA遞交資料,不需向制劑廠商披露,保護了DMF持有人的商業機密;2)對不同制劑廠商引用相同的DMF,只需原料藥廠商向FDA遞交1份資料,FDA也不需重復審評,大大節約了資源;3)制劑廠商在FDA公布的可引用列表中選擇合適的供應商即可,可以將主要精力放在制劑的生產與申報上。由于上述優越性,歐洲及后續的加拿大、澳大利亞等國家的原料藥管理都部分參考了美國DMF制度[3]。本文中對美國DMF注冊申報及年報更新遞交的法規要求進行了研究,以便為國內原料藥管理制度的完善提供借鑒,并為國內制藥企業出口原料藥的國際注冊提供參考[4]。
1 DMF簡介
1.1 概述
DMF是一份向FDA遞交的文件,用于提供生產、包裝、儲存一個或多個人用藥品中的關于廠房設施、工藝流程及所用原料、包裝材料等的保密的詳細信息[5]。沒有任何法規或FDA的規定強制要求企業必須遞交DMF,DMF的遞交完全由申請人自行決定,選擇不遞交DMF而將其作為藥品申請資料的一部分同時申報也是可行途徑。但由于DMF能保證持有人對專有信息(如生產工藝)的保密性,它通常由DMF持有人申請來允許除持有人外的一家或多家企業引用材料,并由FDA對材料進行審查,而不必向用戶披露文件的內容,也不必向每一用戶重復提供資料[6]。
DMF中包含的信息可用來支持臨床研究申請(IND)、新藥申請(NDA)、簡化新藥申請(ANDA)、另一DMF、出口申請及上述任何申請的修訂和補充。若申請者需要引用自身材料,可在上述申請中直接包含這些信息,而不需再創建新的DMF。DMF不能替代IND,NDA,ANDA或出口申請,它不會被批準或否決,僅作為參閱性資料在FDA中心檔案室(CDR)存檔待審。所以DMF本身不存在補充遞交,除原始遞交外只存在修訂和年報(AR)等的遞交。只有當與 IND,NDA,ANDA或出口申請的審核相關聯(即被引用)時,DMF的技術內容才會真正被審核。
2017年4月7日,FDA宣布將以電子通用技術文件(eCTD)格式遞交DMF的合規日期延長至2018年5月5日(原為 2017年 5月5日)。自 2018年 5月5日開始,新遞交的DMF及現有DMF的相關文件,包括本文所討論的DMF年報等,都必須采用eCTD格式遞交。該日期后未使用eCTD格式遞交的DMF相關文件將被拒收。本文中根據eCTD格式標準指出了DMF年報中具體申報文件在遞交申請中應處模塊和部分。
1.2 類型
DMF共分5類。Ⅰ類:涉及生產場地、設施、操作規程及人員,由于這些資料對FDA審評員無直接幫助,FDA于1995年7月3日發布通知正式取消[7],但為了防止混淆,仍保留原DMF分類號;Ⅱ類:涉及原料藥、原料藥中間體及在制備中所用材料,這是數量最多的DMF類型,也是原料藥生產企業出口至美國通常申請注冊的DMF類型;Ⅲ類:涉及包裝材料,藥品包裝材料或組件生產商可以提交該類DMF以保證自身生產信息的保密性;Ⅳ類:涉及藥用輔料、色素、香精、調味料及其他添加劑等非藥性成分;Ⅴ類:包括FDA接受的其他參考資料。若廠家希望遞交Ⅰ~Ⅳ類DMF中未涵蓋的信息,持有人必須向FDA遞交意向書,隨后FDA會聯系持有人。未經與FDA討論而遞交的Ⅴ類DMF將被退回。
1.3 審核
行政審核:當FDA收到包括DMF在內的任何遞交時不會向遞交者發送通知。收到DMF后,原始遞交會進行行政審核以確定遞交在行政上是否完整。行政審核可能持續2~3周。如果從行政角度DMF是可接受的,FDA將發送確認信通知持有人被分配的DMF號。在此時間點DMF狀態變為“激活”。如果從行政的角度DMF不可接受,將通知持有人需要改正的缺陷。DMF原始遞交后,FDA并不再通過郵件或信件確認收到的其余遞交。但實際上所有DMF原始遞交后相關的遞交,包括年報,都會經過行政審核以確定是否完整。如果遞交從行政的角度不可接受,FDA都將通知持有人改正缺陷。DMF的狀態是否為“激活”可在FDA官網的DMF列表中查詢,該列表每季度更新1次[8]。DMF的狀態與該DMF是否經過技術審核或完整性評估無關。在如下2種情形下,DMF的狀態會由“激活”變為“未激活”,一是該DMF由于持有人未在90 d內以年報更新回復逾期通知信(見下文)而由FDA關閉;二是該DMF由DMF持有人申請關閉或撤銷。
技術審核:行政審核完成后,FDA僅將其備案存檔,不會自動啟動技術審核。DMF僅在滿足下列情形后才會進行完整的技術信息審核:1)該DMF狀態為“激活”;2)DMF持有人遞交該DMF的授權信(LOA),如果DMF為CTD格式,無論電子遞交還是紙質遞交,授權信應在“1.4.1”部分遞交,LOA 中應包含 DMF 號;3)持有人將LOA發送給被授權方;4)被授權方向FDA遞交包含LOA 的申請。LOA 應該在申請的“1.4.2”部分遞交。
完整性評估:根據2012年出臺的仿制藥收費法案(GDUFA),于2012年10月1日后首次被引用的支持ANDA的Ⅱ類原料藥DMF需要繳納DMF費,并在付費后進行完整性評估。完整性評估并不能取代完全的科學技術審評,僅確定DMF中包含的信息是否足夠支持ANDA的遞交。通過完整性評估的DMF會被收錄在“可被引用”列表中,該列表每周更新1次[9]。
2 FDA的要求
美國藥品行政法規中并沒有強制DMF必須逐年遞交年報的要求,但DMF相關的行政法規中(d)部分規定,DMF中需包含1份當前授權引用DMF中任何信息的各用戶完整名單[10]。為保證合規,FDA發布的DMF指南建議DMF持有人應當在原始遞交后的每個周年日提供年度報告,而不需每次新增被授權方都提交更新的名單。這構成了遞交DMF年報的法規來源。
DMF指南進一步指出,DMF年報應包含持有人姓名、DMF號、更新日期,注明各個用戶被授權引用信息,可通過日期、卷數、頁數等給出信息的具體位置。DMF年報還應當指出自上次DMF年報后所有變更和增加的信息。如果DMF的內容維持不變,DMF持有人也應當提供1份DMF的內容已為最新版本的聲明。
若DMF持有人未遞交年報或每年向FDA保證之前提交的材料與列表仍是最新的,則相關聯的引用該DMF內容的申請注冊審評也會相應受到影響,FDA可能延遲審查尚未通過的引用該DMF的IND,NDA,ANDA和出口申請或上述任何申請的修訂申請和補充申請。
為確保DMF為最新版本,對于過去36個月中未遞交年報的DMF的持有人,FDA將發送“逾期通知信”。若DMF持有人收到逾期通知信90 d內仍未回復,就遞交年報,FDA將會啟動關閉該DMF的程序。
根據前文對DMF狀態的討論,若FDA關閉該DMF,則FDA定期發布的DMF列表中該DMF的狀態將變為“未激活”。若想再次將DMF的狀態轉回為“激活”,則必須遞交“激活”申請,其中需包含完整的DMF的更新遞交,否則DMF持有人只能再申請新的DMF。
3 遞交實踐
FDA申請遞交的數據庫結構見圖1。其中,修訂分為不同的類別和子類別。對于DMF這一申請類型,下屬遞交類型及相應類別和子類別見表1。
1份DMF年報遞交中包含的文件主要有封面信、行政信息、被授權方列表、修訂遞交記錄等。
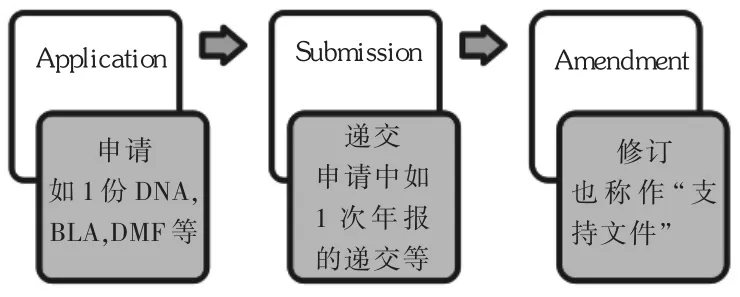
圖1 FDA申請遞交數據庫結構

表1 DMF遞交類型及類別
封面信:DMF年報封面信是對整個遞交材料內容的概述。內容應包括,1)遞交相關基本信息,即遞交日期、DMF類型、DMF號、DMF持有人、主題(即題目,需與FDA官網定期更新的DMF列表中顯示的主題一致);2)封面信抬頭中的遞交類型欄下用粗體注明遞交類型、類別及子類別,DMF年報即為注明遞交類型:年報;3)年報報告期,通常為DMF的原始遞交周年日,跨度為1年;4)正文部分,簡要敘述本次遞交所包含的變更及位于的eCTD模塊和部分位置,正文還應注明經過此次年報更新后該DMF為最新版本,并承諾未來的任何變更會通知FDA及授權用戶;5)持有人或授權代表的簽名,以及簽字人的機打姓名、頭銜、所在單位(持有人或代理人)、電話、傳真、郵箱等。根據eCTD格式要求,該文件在“1.2”部分封面信中遞交。
行政信息:DMF年報中需包含的行政信息與DMF原始遞交中需包含的行政信息一致。包括:1)持有人的姓名和地址;2)生產廠商的名字和地址;3)聯系人的姓名、郵件地址、電話號碼、傳真號碼、電子郵件地址;4)代理人的姓名和地址(如適用);5)聯系人的姓名、郵件地址、電話號碼、傳真號碼、電子郵件地址(如適用)。其中代理人的信息只有當該DMF存在委托代理人時才需遞交,并非必需。根據eCTD格式要求,DMF行政信息在“1.3.1”部分聯系人、申辦方或申請人信息中遞交,其中DMF 持有人及生產廠商的信息更新在“1.3.1.1”部分遞交,代理人及聯系人的信息更新在“1.3.1.2”部分中遞交。
被授權方列表:DMF年報必須包含1份完整的被授權引用該DMF的企業的列表,即使報告期內該列表并沒有發生變化。若沒有被授權方,也必須遞交聲明。該列表指出了當前哪些制劑廠商獲得授權可以引用該DMF 內容。根據 eCTD 格式要求,該文件在“1.4.3”部分被授權引用方列表中遞交。
修訂記錄:DMF年報需列出自上次年報后或原始遞交后(若未遞交過年報)的報告變更的修訂遞交記錄,包括遞交日期、遞交類型及內容的描述。若未遞交任何修訂,也應當遞交聲明。該修訂記錄指出了報告期間原料藥廠商發生了哪些生產變更。根據eCTD格式要求,該文件在“1.13.5”部分生產變更總結中遞交。
根據上述FDA對DMF年報遞交的要求,最終形成的1份DMF年報的完整eCTD結構見圖2。

圖2 1份DMF年報遞交的完整eCTD結構
4 結語
之前,我國一直未將原料藥與制劑加以區別,采用批準文號管理的行政許可制度[11],但目前已逐步在向DMF管理制度過渡。DMF制度是保護知識產權的關鍵機制,但企業遞交完DMF后并不是一勞永逸[12]。藥品監管機構并不會批準DMF,由于原料藥廠商的行政信息、生產信息、被授權人名單等時常會有變更,造成實際情況與申報材料不一致的情形。為了保證當原料藥與制劑關聯進入技術審評時DMF文件為最新版本,DMF持有人需每年定期向FDA遞交更新信息。筆者梳理了FDA對DMF年報遞交的相關規定,并通過實例闡述了合規的DMF年報完整結構和具體內容,對國內企業向國外出口原料藥的DMF注冊申報文件尤其是年報更新的撰寫與遞交具有實際參考價值,同時也對國內原料藥管理向DMF管理制度轉型的管理制度制訂提供一定借鑒。
猜你喜歡
中華手工(2017年2期)2017-06-06 23:00:31
中外會展(2014年4期)2014-11-27 07:46:46
大眾創業(2009年10期)2009-10-08 04:52:00
數字社區&智能家居(2009年7期)2009-09-29 08:16:48
數字社區&智能家居(2009年11期)2009-06-25 04:30:34
數字社區&智能家居(2009年3期)2009-04-21 03:09:04
數字社區&智能家居(2009年2期)2009-03-27 04:33:44
數字社區&智能家居(2009年12期)2009-02-03 07:50:48
建筑創作(2001年3期)2001-08-22 18:48:14
祝您健康(1987年3期)1987-12-30 09:52:32
