分散固相萃取-液相色譜-串聯質譜法測定淡水魚中柱孢藻毒素、節球藻毒素和微囊藻毒素
2019-06-24 08:55:42陳麗惠賈玉珠潘秋仁陳秦秦蔡偉鵬廈門市疾病預防控制中心福建廈門361021
色譜 2019年7期
陳麗惠, 賈玉珠, 張 斌, 潘秋仁, 陳秦秦, 蔡偉鵬(廈門市疾病預防控制中心,福建 廈門 361021)
藍藻毒素是藍藻代謝過程中產生的有毒次生代謝產物,按照毒素作用的靶器官分類,主要有肝毒素、神經毒素、皮膚毒素和細胞毒素等[1]。其中,藍藻肝毒素包括柱孢藻毒素(CYN)、節球藻毒素(NOD)和微囊藻毒素(MCs)3大類,常見的微囊藻毒素有MC-RR、MC-YR及MC-LR。藍藻肝毒素不僅直接污染水體[2,3],還能在淡水生物體內產生蓄積[4,5],再通過食物鏈進入人體,導致肝臟和腎臟損傷[6],危害公眾健康。因此,建立快速、靈敏、簡便、可靠的藍藻肝毒素的檢測方法對于保障人民飲食安全具有重要意義。
目前,國內外的柱孢藻毒素、節球藻毒素和微囊藻毒素的檢測方法主要有液相色譜法(LC)[7,8]、液相色譜-串聯質譜法(LC-MS/MS)[9-12]和鳥槍串聯質譜法[13]。已見報道的研究主要針對單類藍藻毒素,未見同時檢測CYN、NOD和MCs的文獻報道。魚肉中的CYN、NOD和MCs的前處理方法主要采用固相萃取法[14-16],但其過程繁瑣,消耗溶劑大,分析成本偏高。
分散固相萃取法(DSPE)是2003年美國農業部提出的一種操作簡單、快速、成本低廉的樣品前處理技術[17],其原理是通過將吸附劑加入到樣品提取液中,吸附干擾雜質,以達到凈化樣品的目的。對比傳統的固相萃取技術,該技術更為簡便,有效提高了前處理效率,并避免固相萃取中柱子易堵塞的問題,近年來在食品、環境等領域得到了廣泛應用[18-20]。目前,以分散固相萃取法對CYN、NOD和MCs同時進行提取劑凈化的方法未見報道。
本研究采用分散固相萃取法對魚肉進行樣品前處理,著重考察了提取溶劑及吸附劑的種類對提取效率及凈化效果的影響,結合液相色譜-串聯質譜技術,對淡水魚中CYN、NOD及MCs(MC-RR、MC-YR和MC-LR)進行同時檢測。該方法可為淡水魚中多種藍藻肝毒素的快速測定提供可靠的技術支持。
1 實驗部分
1.1 儀器、試劑與材料
6460液相色譜-三重四極桿質譜儀、1260高效液相色譜儀(美國Agilent公司);冷凍高速離心機(德國Sigma公司);KQ-500E型超聲波清洗機(昆山市超聲儀器有限公司)。
CYN、NOD及MCs(MC-RR、MC-YR、MC-LR)標準品由北京曼哈格生物科技有限公司提供;乙腈和甲醇(色譜純,德國Sigma公司);N-丙基乙二胺粉末(PSA,天津博納艾杰爾科技有限公司);C18粉末和酸性氧化鋁粉末(A-AL)(美國Agilent公司);氯化鈉(國藥集團化學試劑有限公司)。
取適量上述標準溶液,用甲醇配制5種目標化合物的混合標準溶液,其中MC-RR、MC-YR、MC-LR及NOD的質量濃度為0.4 mg/L,CYN為1.0 mg/L,置于-18 ℃下避光保存。吸取適量上述混合標準溶液,用空白基質提取液稀釋定容,配制成不同濃度的基質匹配標準溶液。
1.2 樣品前處理
稱取2.0 g(精確至0.01 g)粉碎均勻的市售鰱魚魚肉樣品,置于30 mL離心管中,加入10 mL乙腈-水-甲酸溶液(89∶10∶1,v/v/v),渦旋振蕩提取3 min,以10 000 r/min低溫離心10 min,取2.0 mL上層清液,置于15 mL玻璃離心管中,加入100 mg C18吸附劑,渦旋混勻1 min,靜置分層,取1.0 mL上清液,加入2.0 mL水,混勻后,過0.22 μm有機濾膜,待上機分析測定。
1.3 色譜條件
Agilent ZORBAX Eclipse XDB C18色譜柱(50 mm×4.6 mm,1.8 μm);柱溫為35 ℃;流動相A為水,B為乙腈;流速為0.3 mL/min。梯度洗脫程序為0~1.0 min,10%B~70%B;1.0~5.0 min,70%B~90%B;5.0~9.0 min,90%B;9.0~9.1 min,90%B~10%B;9.1~15.0 min,10%B。進樣體積:10 μL。
1.4 質譜條件
離子源:ESI源,負離子模式;氣流溫度:350 ℃;氣流速度:8 L/min;霧化氣壓力:0.2 MPa;鞘氣(N2)溫度:350 ℃;鞘氣(N2)流速:11 L/min;毛細管電壓:4 500 V;噴霧電壓:0 V,電子倍增電壓:580 V。其他質譜參數見表1。

表1 柱孢藻毒素、節球藻毒素、微囊藻毒素-RR、微囊藻毒素-YR和微囊藻毒素-LR的質譜參數Table 1 MS parameters of cylindrospermopsin (CYN),nodularin (NOD),microcystin-RR (MC-RR),microcystin-YR (MC-YR)and microcystin-LR (MC-LR)
* Quantitative ion.
2 結果與討論
2.1 質譜和色譜條件的優化
MCs和NOD在結構上為環狀多肽,CYN是一種環類生物堿,均為兩性化合物,在ESI(+)和ESI(-)兩種掃描模式下進行質譜分析時,均能找到相應的母離子,因此有必要對其響應強度進行比較,以選擇合適的離子源模式。本實驗考察了1.0 mg/L的5種藍藻肝毒素的標準溶液在ESI(+)和ESI(-)模式下待測物的離子響應。以乙腈-0.1%(v/v)甲酸水溶液為流動相,考察5種待測物在ESI(+)模式下的響應,NOD和MC-RR的響應較好,CYN、MC-YR和MC-LR的響應較差。同時考察了在ESI(-)掃描模式下以乙腈-水溶液為流動相時待測物的響應,CYN、NOD、MC-YR和MC-LR的離子化效率較在ESI(+)模式下高,但MC-RR的質譜響應較差。綜上,選擇的離子化模式為ESI(-),與文獻[21-23]報道相符。
在ESI(-)模式下,以乙腈-水為流動相,對待測物進行全掃描以確定各自的母離子,在二級質譜掃描中選擇豐度最高的子離子作為定量離子,豐度次之的為定性離子,并對碰撞能量進行優化,具體質譜參數見表1。
分別選用Waters BEH C18(50 mm×2.1 mm,1.7 μm)、Phenomenex Luna C18(150 mm×2.0 mm,3 μm)和Agilent ZORBAX Eclipse XDB C18(50 mm×4.6 mm,1.8 μm)色譜柱進行分析,以對色譜條件進行優化。采用Phenomenex Luna C18色譜柱時,MC-RR、MC-YR和MC-LR存在拖尾現象;采用Waters BEH C18色譜柱時,目標化合物的峰形較差;采用Agilent ZORBAX Eclipse XDB C18色譜柱時,MC-RR、MC-YR和MC-LR的色譜峰形及分離度最佳,且與CYN和NOD實現完全分離。因此,選取Agilent ZORBAX Eclipse XDB C18色譜柱對5種藍藻肝毒素進行分離。
考察了采用乙腈-水,乙腈-5 mmol/L乙酸銨、乙腈-0.1%(v/v)甲酸水溶液作為流動相時的分離效果及離子化效率。結果表明,使用乙腈-水為流動相時,5種目標化合物的質譜響應較高,色譜峰形對稱尖銳。因此本研究選擇乙腈-水作為流動相。圖1為基質加標樣品中5種目標化合物的提取離子色譜圖,目標物出峰段雜質干擾較小。
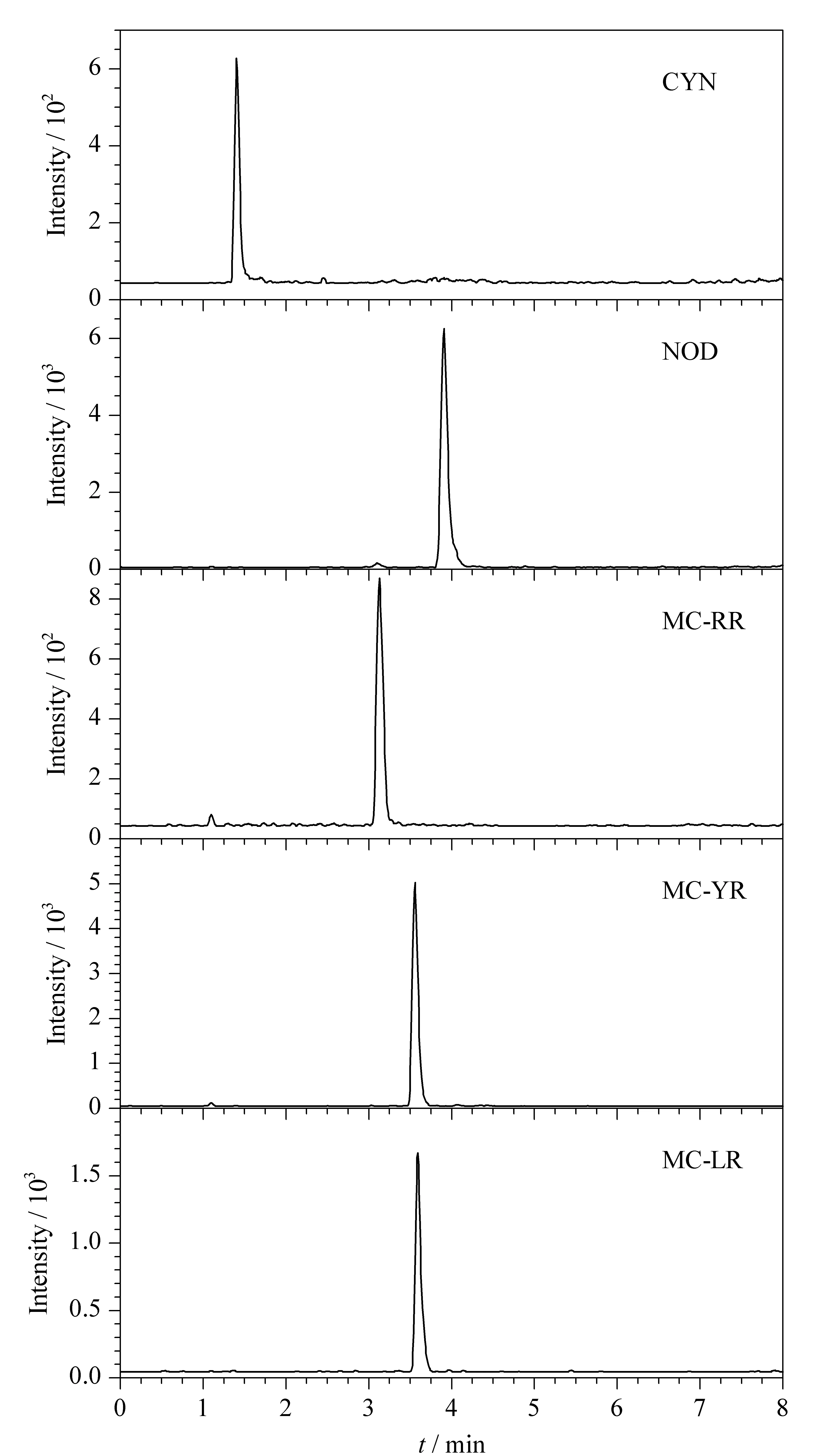
圖1 基質加標樣品中5種化合物(5 μg/L)的提取離子色譜圖Fig.1 Extracted ion chromatograms of the five compounds (5 μg/L)in matrix spiked samples

圖2 不同提取溶劑對5種化合物回收率的影響(n=3)Fig.2 Effect of different extraction solvents on the recoveries of the five compounds (n=3) A:methanol-formic acid (99∶1,v/v);B:acetonitrile-water (90∶10,v/v);C:methanol;D:acetonitrile-water-formic acid (89∶10∶1,v/v/v).
2.2 分散固相萃取前處理條件優化
2.2.1提取溶劑的選擇
甲醇、甲醇水溶液和乙腈水溶液是提取生物體中藍藻毒素常用的提取溶劑[24-26],在提取溶液中加入適量甲酸可提高藍藻毒素的提取效率[27]。因此,本研究考察了采用甲醇、甲醇-甲酸(99∶1,v/v)、乙腈-水(90∶10,v/v)和乙腈-水-甲酸(89∶10∶1,v/v/v)時的提取效率(見圖2)。結果表明,以乙腈-水-甲酸(89∶10∶1,v/v/v)為提取液時,5種目標化合物的回收率為66.0%~97.4%,明顯高于采用乙腈-水(90∶10,v/v)為提取液時的14.1%~47.9%和采用甲醇-甲酸(99∶1,v/v)時的61.4%~112.4%,也高于甲醇的22.7%~43.7%。實驗結果表明,在提取液中加入1%(v/v)甲酸可明顯提高待測物的提取效率,并有效降低基質效應。綜合考慮,實驗選擇以乙腈-水-甲酸(89∶10∶1,v/v/v)為提取溶劑。
2.2.2進樣溶劑的優化
本方法以乙腈-水-甲酸(89∶10∶1,v/v/v)作為提取液,發現提取液提取樣品后直接進樣,基質效應較大,且由于進樣溶液與流動相初始比例相差較大,大部分化合物的峰形較差。因此,為減少基質效應,同時優化峰形,可在提取液中加入水進行稀釋[26]。本實驗采用在1.0 mL凈化液中加入2.0 mL水進行稀釋,使進樣溶劑與初始流動相乙腈-水體系更接近,以優化峰形。
2.2.3分散固相萃取吸附劑的選擇
分散固相萃取的吸附劑應吸附提取液中的主要雜質但不吸附待測物,以達到凈化提取液的目的。C18、PSA和A-AL是常見的分散固相萃取吸附劑,用于吸附魚肉及其他食品中的多種干擾雜質,起到凈化樣品提取液的作用[28-31]。其中,C18為非極性吸附劑,主要去除脂類和弱極性雜質;PSA為弱陰離子交換劑,主要去除有機酸、金屬離子和酚類等[32];A-AL也有一定的脫脂效果,能夠降低基質干擾[33]。本實驗比較了C18、PSA和A-AL 3種分散固相萃取吸附劑對魚肉樣品提取液中待測物的凈化回收效果(見圖3)。以A-AL和PSA為吸附劑時,5種待測物的回收率均低于60%,達不到檢測要求。以C18為吸附劑對魚肉樣品提取液進行凈化時,凈化效果及回收率明顯優于其他2種吸附劑。因此,本研究選用C18作為分散固相萃取的吸附劑。
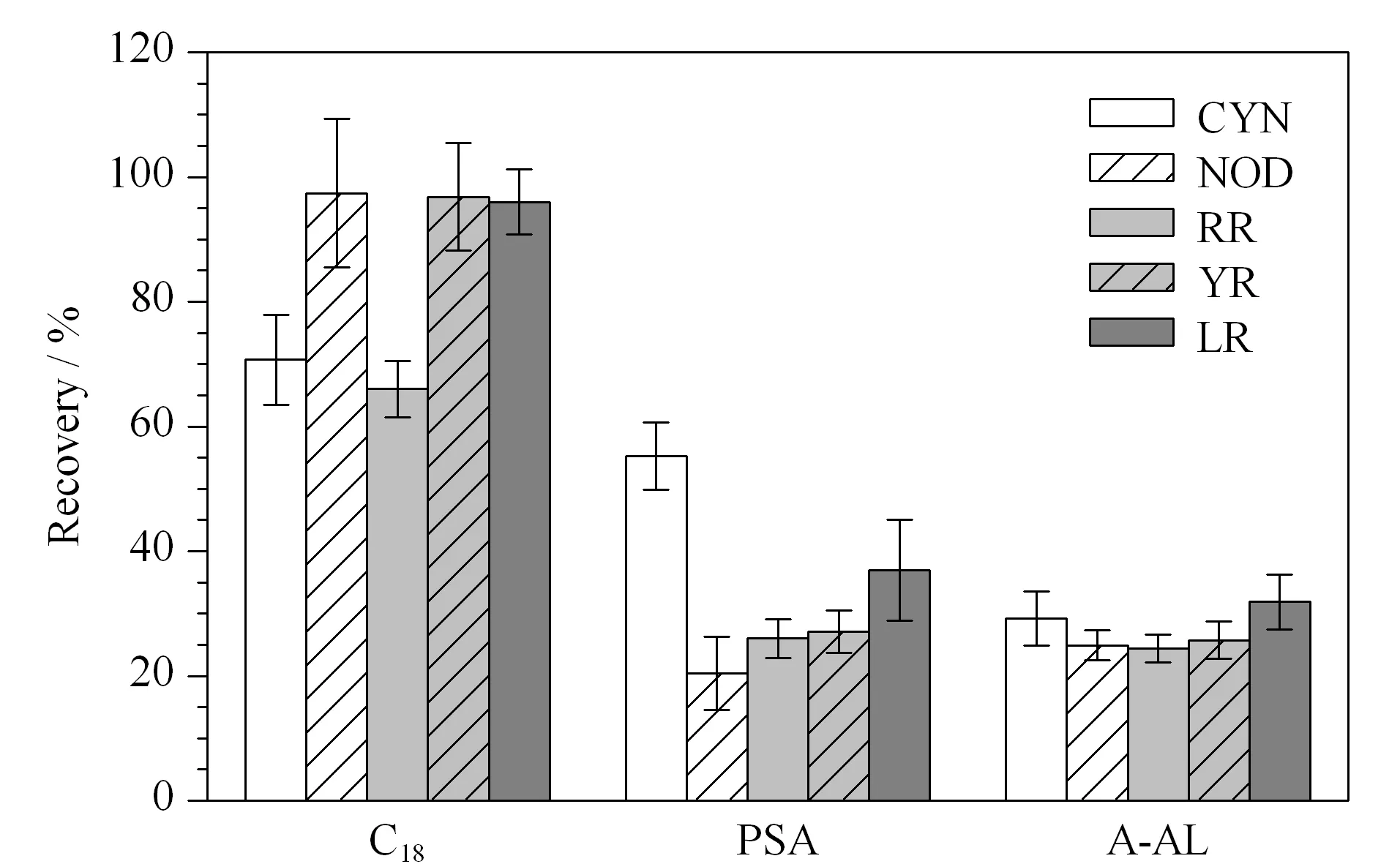
圖3 不同吸附劑對5種化合物回收率的影響(n=3)Fig.3 Effect of different sorbents on the recoveries of the five compounds (n=3)PSA:primary secondary amine;A-AL:acidic alumina.
2.3 基質效應的評估
用乙腈-水-甲酸(89∶10∶1,v/v/v)作為稀釋液配制溶劑標準曲線,同時取不含待測物的魚肉樣品,按照前處理步驟進行提取、凈化,得到空白樣品基質溶液,以空白樣品基質溶液為稀釋液配制基質校準曲線。根據公式,基質效應=基質匹配校準曲線的斜率/溶劑標準校準曲線的斜率[34],斜率在0.8~1.2之間可認為無基質效應,斜率越接近1,表明基質效應越弱。實驗結果表明,5種待測物的斜率比均低于0.8,表明樣品經過分散固相萃取后雖然取得了良好的凈化效果,但仍無法完全消除基質效應,而基質效應的存在會影響檢測結果的準確性,為消除基質效應干擾,實驗采用相應基質匹配標準溶液進行校準。
2.4 方法評價
2.4.1線性范圍、檢出限和定量限
用空白基質提取液作為稀釋液,配制5種目標化合物的基質標準溶液,經過LC-MS/MS測定后,以目標化合物定量離子的峰面積(y)為縱坐標,質量濃度(x,μg/L)為橫坐標作圖,繪制基質校正標準曲線。結果表明,5種目標化合物在各自范圍內線性關系良好,相關系數(R2)為0.995 4~0.999 4。以3倍和10倍信噪比計算得5種目標化合物的檢出限(LOD)和定量限(LOQ),結果表明,LOD為5~10 μg/kg,LOQ為15~40 μg/kg(見表2)。
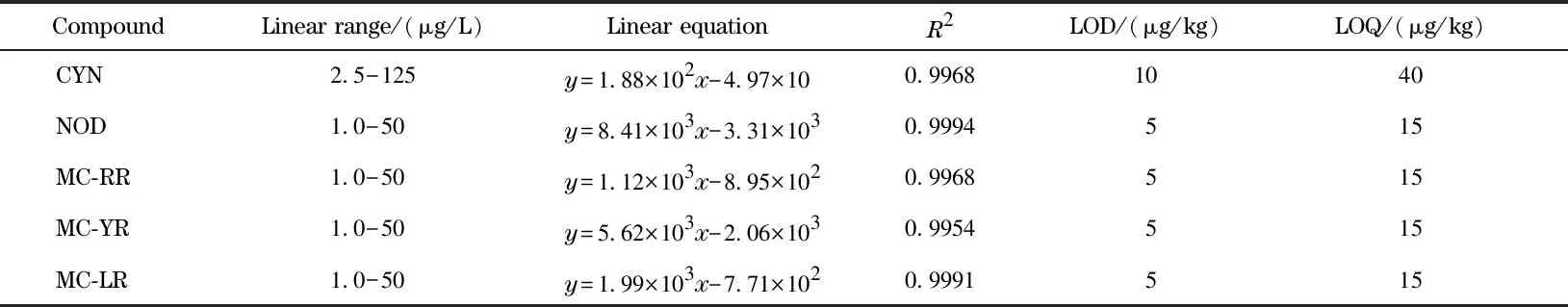
表2 5種目標化合物的線性范圍、線性方程、相關系數、檢出限和定量限Table 2 Linear ranges,linear equations,correlation coefficients (R2),limits of detection (LODs)and limits of quantification (LOQs)
y:peak area of quantitative ion;x:mass concentration,μg/L.
2.4.2回收率和精密度
在陰性鰱魚肉樣品中進行加標回收試驗和精密度驗證。在粉碎后的陰性鰱魚肉樣品中,NOD、MC-RR、MC-YR、MC-LR的低、中、高添加水平為30、60和150 μg/kg,CYN的低、中、高添加水平為75、150和375 μg/kg,按照1.2節進行樣品前處理,再進行LC-MS/MS測定,5種目標化合物的回收率為62.3%~101.2%,RSD(n=6)為0.3%~8.6%(見表3)。

表3 5種目標化合物的平均回收率與精密度(n=6)Table 3 Average recoveries and precisions of the five target compounds (n=6)
Low,medium and high levels:30,60 and 150 μg/kg for NOD,MC-RR,MC-YR and MC-LR;75,150 and 375 μg/kg for CYN.
2.5 實際樣品分析
采用本方法對市售11份鯽魚、10份草魚、6份鰱魚及3份野生鯽魚的魚肉樣品中CYN、NOD及MCs(MC-RR、MC-YR、MC-LR)的含量進行檢測。結果表明,所有樣品的魚肉均未檢出CYN、NOD及MCs。
3 結論
本研究建立了分散固相萃取-液相色譜-串聯質譜同時測定淡水魚中柱孢藻毒素、節球藻毒素和微囊藻毒素的方法。與現有方法相比,該法樣品前處理更為簡便、高效、經濟,適用于淡水魚中多種藍藻肝毒素的快速篩查和準確定量。
