2-氨基苯腈合成拉帕替尼藥物中間體工藝
2019-07-02 12:12:36王艷戎李子成
實驗室研究與探索 2019年6期
關鍵詞:體系
張 宏, 王艷戎, 羅 蘭, 李子成
(1.四川化工職業技術學院 化學工程系,四川 瀘州 646099;2.瀘州職業技術學院 科技產業處,四川 瀘州 646000;3.四川大學 化工學院, 成都 610065)
0 引 言
N-(3-氯-4-(3-氟芐氧基)苯基)-6-溴喹唑啉-4-胺是靶向抗腫瘤藥物拉帕替尼(Lapatinib)的重要中間體之一[1-2]。Lapatinib是由GSK公司開發研制,經全球眾多研究機構近年來的努力,拉帕替尼的合成工藝正向著綠色、環保、高效、可循環的方向發展[3]。本文通過優化文獻方法,以2-氨基苯腈為起始原料,經碘代、縮合、Dimroth重排閉環反應,制備N-(3-氯-4-(3-氟芐氧基)苯基)-6-溴喹唑啉-4-胺,其結構經LC-MS、1H-NMR確定,同時對相關碘代試劑的反應條件進行評價,增強2-氨基苯腈碘化效果,提高2-氨基-5-碘苯腈收率。
1 實驗部分
1.1 主要設備和試劑
設備:LCMS-液質聯用儀(Shimadzu 2010A,島津)。
試劑:2-氨基苯腈(99.5%,上海氟德化工);間氟芐基氯(99.5%,阿法埃莎(中國)化學有限公司);2-氯-4-硝基苯酚(99.0%,百靈威科技有限公司);一氯化碘(99.0%,上海邁瑞爾化學技術有限公司);N,N-二甲基甲酰胺二甲縮醛(99.3%,北京欣賽維化學科技有限公司);薄層層析用硅膠(GF254,青島海洋化工廠);其余試劑均為阿拉丁試劑,使用前經過二次除水(必要時)。
1.2 N-(3-氯-4-(3-氟芐氧基)苯基)-6-溴喹唑啉-4-胺合成路線
Williamson醚化反應是該系列反應的關鍵步驟之一。相關研究顯示,化合物(2)3-氯-4-(3-氟芐氧基)苯胺是以間氟芐醇或鹵代間氟芐基苯為初始原料,首先在堿性體系下,以KI為催化劑,通過Williamson縮合成醚。堿性試劑可以選用有機堿(三乙胺、二異丙基乙胺、二乙胺)和無機堿(Na2CO3、NaHCO3、KHCO3、K2CO3),以收率作為評價指標的單因素實驗中發現,K2CO3-KI體系下的產物(1)收率最高,副反應較少,因此堿性試劑選用K2CO3。溶劑體系可以選用DMF、CH3CN、丙酮、乙酸乙酯。但DMF沸點較高,常規蒸餾和減壓旋蒸都不易回收循環利用;CH3CN體系雖成醚效果較好,但毒性較大、成本昂貴;乙酸乙酯體系的成醚效果較差;丙酮體系對無機堿性催化體系(K2CO3-KI)的溶解效果優于乙酸乙酯,成醚效果較好,且價格便宜,沸點低,回收精制后可循環使用[4-5]。
隨后將硝基(—NO2)加氫還原成胺基(—NH2)。還原試劑可采用H-試劑(NaH)、催化加氫(Pd/C)、金屬還原(Fe粉/NH4Cl)等多種方法,由此可見還原劑的選擇及活性是化合物(2)合成的關鍵點。NaH易燃且不易保存;貴金屬Pd/C催化劑成本較高,并且需要肼鹽做供氫體;Fe/NH4Cl還原反應后會產生大量鐵泥,后處理工藝繁瑣。為此,選擇活性較高、易后處理的Zn/NH4Cl還原硝基的體系。
從經濟高效、適宜工業生產的角度出發,結合眾多學者研究成果,化合物(2)的合成途徑[6-7]:以2-氯-4-硝基苯酚和間氟芐基氯為初始原料,K2CO3-丙酮體系下進行Williamson成醚反應生成化合物(1)3-氯-4-(3-氟苯甲氧基)硝基苯,在Zn/NH4Cl還原體系下,由化合物(1)制備得到化合物(2)。
化合物(4)合成途徑:以2-氨基苯腈為原料,ICl為碘化劑合成化合物(3)2-氨基-5-碘苯腈,于DMF-DME體系下生成化合物(4)N′-(4-碘-2-氰基苯基)-N,N-二甲基甲酰胺[9-10]。
一般而言,碘代過程通常使用高價碘代試劑(ICl、N-碘代丁二酰亞胺、碘乙酸等),無需添加額外的助氧化劑,但碘化過程收率并不理想,碘代試劑通常具有高毒、高腐蝕性,對環境弊端較大。根據綠色環保的化學理念,眾多研究者對碘代-催化體系、助氧化劑、溶劑進行多方面的探討[11,13],如I2-H5PV2MO10O40-O2/CH3CN-HOAc碘化體系;KI(KIO3)/NaNO2/H2SO4-CH3CN碘化體系;I2-H2O2-磺化MCM-41介孔分子篩/HCl碘化體系;NH4I/H2O2/PhMe碘化體系等。在以ICl為碘代試劑進行反應的同時,對I2-O2/Zn(NO3)2·6H2O/H2O碘化體系,I2/NO2/乙酸乙酯-乙腈碘化體系,NH2I/H2O2碘化體系進行評價(以2-氨基-5-碘苯腈的轉化率、選擇性為主要指標)。
化合物(2)、(4)通過Dimroth 重排閉環反應,得到目標產物化合物(5),合成過程無SOCl2、POCl3、水合肼等嚴重污染的毒性物質,更加安全環保,適宜一鍋法反應。所設計的合成途徑如圖1所示。
1.3 HPLC檢測方法
色譜柱Ultimate?XB-C18;流動相[12]:V(pH4.0乙酸銨緩沖液∶CH3CN)=70∶30;流速1.0 mL/min;檢測波長260 nm;柱溫35 ℃;進樣量15 μL。
2 合成方法與結果
2.1 化合物(1)的制備
在150 mL三口燒瓶中,加入2.60 g(15 mmol)2-氯-4-硝基苯酚,2.85 g(18 mmol)K2CO3,0.30 g(1.8 mmol)KI,75 mL丙酮,開啟攪拌,15 min后加入2.16 g(15 mmol)間氟芐基氯,加熱回流,TLC點板監控反應進程(展開體系V(乙酸乙酯∶石油醚)=1∶2),6.5 h后停止加熱,自然降溫過濾,丙酮洗滌濾餅3次(20 mL/次),合并濾液,減壓蒸干溶劑,真空干燥,得到淺黃色固體4.01 g,HPLC檢測純度95.2%,收率92.0%。LC-MS:283[M+H]+,1H-NMR (DMSO,300 MHz):8.36(d,J=2.6 Hz, 1H), 8.25(J=9.2 Hz, 2.7 Hz, 1H), 7.42(m, 2H), 7.33(m, 2H), 7.24(t, 1H), 5.43(s, 2H)。

圖1 N-(3-氯-4-(3-氟苯甲氧基)苯基)-6-碘喹唑啉-4-胺合成路線
2.2 化合物(2)的制備
Zn粉活化:稱取10 g未活化鋅粉置于50 mL 18 mol/L的鹽酸溶液中,玻璃棒快速攪拌3 min后,混合液轉移至G3砂芯漏斗內抽濾,蒸餾水洗滌濾餅至中性,CH3CH2OH洗滌2次,將濾餅置于真空干燥箱,25 ℃干燥4 h。活化后的Zn粉不宜長時間保存,一般使用前取相應量進行活化。
化合物(2)制備:1.2 g NH4Cl溶于少量水中,與50 mL乙醇混合后在攪拌條件下加入7.2 g活化后的Zn粉,用10%的鹽酸調節體系pH至4,加熱至45 ℃,攪拌30 min后,分3次(1.00 g/次)加入化合物(1),加熱回流,TLC點板監測反應進程,2~3 h即可完全反應。趁熱過濾,乙醇洗滌濾餅3次(20 mL/次),合并濾液,50 mL飽和食鹽水反洗有機相,CH2Cl2-乙酸乙酯萃取,收集有機相,減壓旋干,固相真空干燥,得到黃色固體2.48 g,HPLC檢測純度93.6%,收率86.9%。LC-MS:253[M+H]+;1H-NMR (DMSO, 300 MHz): 7.47~7.35(m, 1H), 7.29~7.24(m, 2H), 7.18~7.12(t, 1H), 6.93(d,J=8.6 Hz, 1H), 6.64(d,J=2.7 Hz, 1H), 6.49~6.44(J=8.7Hz, 2.5Hz, 1H), 5.08(s, 2H), 4.95(s, 2H)。
2.3 化合物(3)的制備
2.3.1 ICl-HOAC碘代體系
150 mL燒瓶內加入3.54 g(30 mmol)2-氨基苯腈完全溶于50 mL HOAc。將5.00g(30.8 mmol)ICl溶于20 mL HOAc中,在N2保護下,通過恒壓滴液漏斗緩慢滴入燒瓶內,室溫攪拌,4.5 h停止反應,向反應液中加入150 mL飽和食鹽水,過濾,蒸餾水洗滌濾餅3次。濾餅于真空干燥箱干燥后,苯/石油醚(V=1∶3)體系下進行重結晶,得到3.92 g片狀晶體,純度90.3%,轉化率48.2%。1H-NMR (DMSO, 300 Hz):7.71(d,J=2.2 Hz, 1H), 7.55(dd, 1H), 6.65(d,J=8.7 Hz, 1H), 6.24(s, 2H)。
2.3.2 I2-O2/Zn(NO3)2·6H2O/H2O碘化體系(Ⅰ)
150 mL燒瓶內加入30 mmol 2-氨基苯腈,5 mmol Zn(NO3)2·6H2O,15 mmol單質I2,60 mL H2O。首先在N2保護下[14-15],攪拌加熱至有回流產生,將N2切換至O2-N2(體積比4∶1)混合氣體,反應過程中通過HPLC對化合物(3)的轉化情況進行跟蹤分析。反應結束后,加入適量6%硫代硫酸鈉水溶液進行反應淬滅,反應液中加入150 mL飽和食鹽水,過濾,將濾餅溶于HOAc中,適量蒸餾水反洗有機相,分液,將HOAc旋蒸脫除,苯/石油醚(V=1∶3)體系下進行重結晶片狀結晶,化合物(3)的轉化率、目標產物選擇性隨反應時間的變化見表1。由表1數據可知,0~10 h內化合物(3)的轉化率快速增長至44.7%,10~20 h化合物(3)轉化率增長幅度降低,隨著反應時間的進行,副反應增多,產物的選擇性逐步降低。此反應體系與ICl-HOAc碘化體系對比,化合物(3)轉化率沒有明顯提升,反應時長增加約4.3倍,故此碘化體系不太適用于2-氨基苯腈的碘化反應。

表1 碘化體系(Ⅰ)反應情況
O2作為助氧化劑與I2進行碘化反應,其機理可能是O2/Zn2+/硝酸根協同促使單質I2形成I8+高價碘活性中間體[16],如圖2所示。
2.3.3 I2/NO2/乙酸乙酯-乙腈碘化體系(Ⅱ)
為便于實驗操作和NO2計量,從安全角度出發,將NO2通入(質量分數)15%NaOH溶液中,制備6.5 mol%的氮氧化物鈉鹽,進行后續的碘化反應。
2NO2+2NaOH?NaNO3+NaNO2+H2O

圖2 碘化過程機理推測
向250 mL帶聚四氟乙烯內襯的高壓反應釜內加入30 mmol 2-氨基苯腈,15 mmol單質I2,80 mL 6.5 mol%的NO2-NaOH溶液,30 mL CH3CN-HOAc混合溶劑(體積比3∶1)。反應釜首先用N2置換3次,之后通入N2至壓力達到0.5 MPa,攪拌加熱至有回流產生,將N2切換至O2-N2(體積比4∶1)混合氣體,反應過程中通過HPLC對化合物(3)的轉化情況進行跟蹤分析,化合物(3)的轉化率、選擇性隨反應時間的變化見表2。
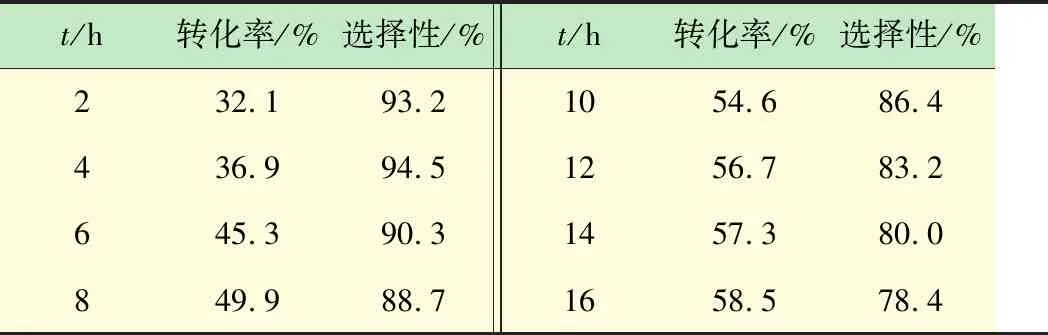
表2 碘化體系(Ⅱ)反應情況
由表2數據可知,反應8 h化合物(3)的轉化率達到49.9%,反應16 h后轉化率達到58.2%,雖然耗時較高,但相比于ICl體系,轉化率增長了10%左右。同樣,反應時間的延長,副反應增多,生成產物的選擇性逐步降低。從綠色環保反應角度出發,碘化體系(Ⅱ)的環境負荷低于ICl-HOAc。
2.3.4 NH2I/H2O2碘化體系(Ⅲ)
150 mL燒瓶內加入30 mmol 2-氨基苯腈,80 mL甲苯。開啟攪拌至氨基苯腈完全溶解,在加入30 mmol NH2I,在N2保護下,加熱至產生回流,將300 mmol H2O2通過恒壓滴液漏斗勻速滴入燒瓶內,滴加完畢后,繼續在N2保護情況下進行碘化反應。反應過程中通過HPLC對化合物(3)的轉化情況進行跟蹤分析,其轉化率、選擇性隨反應時間的變化見表3。
由表3數據可知,反應4h化合物(3)的轉化率僅有9.4%,反應66h后轉化率達到47.8%,與ICl-HOAc體系相似,反應78h,化合物(3)的轉化率基本穩定在63%左右,此體系碘化反應時間很長,相比于ICl體系,轉化率增長了15%左右,但過程的副反應較少,90 h的產物選擇性還在90%以上。就綠色化學而言,碘化體系(Ⅲ)的環境負荷顯著低于ICl-HOAc,碘化反應中催化劑種類較少,反應條件溫和易達到,如果能有效降低碘化反應所需時間,該路線基本符合工業化大生產的要求。

表3 碘化體系(Ⅲ)反應情況
2.4 化合物(4)的制備
將2.20 g化合物(3)加入至6 mL N,N-二甲基甲酰胺二甲基縮醛(DMF-DMA)中,加熱回流1.5 h,減壓蒸出過量的DMF-DMA,得到化合物(4)直接進行下一步Dimroth重排閉環反應。
2.5 化合物(5)的制備
將化合物(4),2.0 g(7.97 mmol)化合物(2),20 mL HOAc加熱回流1.5 h后,停止加熱,自然降溫。60 mL飽和食鹽水反洗反應液,CH2Cl2-乙酸乙酯萃取,合并有機相,旋蒸除溶劑,固相經真空干燥,得到黃色固體3.58 g,HPLC檢測純度91.0%,收率81.4%。LC-MS:507[M+H]+;熔點:222.0~224.0 ℃,1H-NMR(DMSO, 300 Hz):9.86(s, 1H), 8.97(s, 1H), 8.65(s, 1H), 8.16(d,J=8.7 Hz, 1H),8.08(s,1H),7.79(d,J=9.0 Hz,1H),7.57(d,J=8.6 Hz, 1H), 7.48(m,1H), 7.35~7.27(m, 3H), 7.21(t, 1H), 5.30(s, 2H)。
3 結 語
本文以2-氯-4硝基苯酚和間氟芐氯通過Williamson縮合成醚,Zn/NH4Cl還原制備化合物(2),反應的總收率為79.9%。此步驟的還原體系Zn/NH4Cl,Zn粉經過活化處理,提高了還原效率。該體系較Pd/C體系、Raney Ni-水合肼體系、NaH體系相比,在盡量保證產品收率的前提下,降低生產成本,未經活化的Zn/NH4Cl易于長期保存。
以ICl為碘化劑,對2-氨基苯氰進行進行碘代反應合成化合物(3),化合物(3)在DMF-DME體系下進行胺-醛縮合得到化合物(4),化合物(4)與化合物(2)在HOAc體系下進行Dimroth 重排閉環反應制得目標產物化合物(5),單步收率84.1%。該過程為拉帕替尼制備過程中的Suzuki偶聯反應提供原料,基本符合投入工業化生產的要求。
然而,反應過程中應用的碘代劑ICl對環境有一定的負荷,遇水易分解,碘代效果不理想,故由2-氨基苯氰制備化合物(5)反應總收率僅為40.5%。因此對三種新碘化體系進行評價:I2-O2/Zn(NO3)2·6H2O/H2O碘化體(Ⅰ)系,I2/NO2/乙酸乙酯-乙腈碘化體系(Ⅱ),NH2I/H2O2碘化體系(3)。碘化體系(Ⅰ)不適合2-氨基苯氰的碘化反應;碘化體系(Ⅱ)的產物轉化率有所提升,但選擇性不理想;碘化體系(3)反應耗時較長,但產物的轉化率和選擇性較為理想,反應90 h,2-氨基苯腈轉化率63%,選擇性91.2%,較為符合綠色化學的要求。
猜你喜歡
商品與質量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛生(2015年12期)2015-11-10 05:13:40
現代企業(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
