橄欖油中農藥多殘留前處理方法研究及其應用于氣相色譜、氣質聯用測定效果評估
2019-07-17 03:35:54蘇建峰陳勁星連文浩劉建軍
中國糧油學報 2019年6期
蘇建峰 陳勁星 連文浩 馬 瑩 徐 佳 沈 松 劉建軍,
(福建中檢華日食品安全檢測有限公司1,福州 350008) (空軍第一后勤訓練基地2,上海 200120)(中國檢驗認證集團福建有限公司3,福州 350015)
橄欖油是由新鮮的油橄欖果實冷榨而成,不經加熱和化學處理,保留了天然營養成分,營養價值高。由于橄欖在種植和生長過程中使用農藥用于防治蟲害,且部分農藥不易降解,致使橄欖油中可能存在農藥殘留。橄欖油中含有大量油脂,基質干擾大,屬于較難分析的樣品類型。由于農藥多殘留技術[1-10]監測項目范圍廣,優勢顯著,近年來已有不少植物油中農藥多殘留分析的研究報道[11-20],主要采用凝膠滲透色譜[13-15](GPC)凈化、色譜質譜聯用分析,GPC在油脂凈化方面效果較好,但需要專門設備,并且效率不高;其他如冷凍、分散固相萃取法等[16-17]快速法效率較高,但對油脂中大量干擾基質的凈化效果一般,常需配合二級串聯質譜(LC-MS/MS、GC-MS/MS)的強抗干擾性進行分析,且基質效應較強影響定量結果;傳統的液液分配法、固相萃取法等[18-19]多用于分析某一兩類(如有機磷類、菊酯類等)農藥幾種或幾十種,適用范圍較窄。
本研究建立并優化串聯雙柱凈化方案,用于橄欖油中農藥多殘留分析前處理,操作迅速、適用項目范圍遠超已有的文獻報道(在進出口農藥篩查品種的基礎上進一步擴充,跨種類達313種農藥),并且項目列表中包含了多殘留分析技術較難并入的部分農藥如:乙酰甲胺磷、百菌清等。在儀器測試方面,考察了該前處理凈化方案在不同儀器上的效果,結果表明,各儀器的譜圖均呈現較優的凈化效果,可依據具體情況選擇配合GC-MS、GC-ECD或GC-FPD進行測定,實用性強,滿足高通量樣品的檢測需求,文中對實驗的前處理和儀器測定參數進行了探討。
1 試驗部分
1.1 儀器與試劑
7890A-5975C氣相色譜-質譜聯用儀,7683自動進樣器,EI源;Clarus 680氣相色譜儀,ECD檢測器,FPD檢測器;色譜柱:DB-5MS毛細管柱30 m×0.25 mm×0.25 μm,HP-5毛細管柱30 m×0.32 mm×0.25 μm,DB-1701毛細管柱30 m×0.32 mm×0.25 μm;313種農藥標準品(逐級稀釋后備用);Envi-18、PSA固相萃取小柱。
1.2 實驗方法
1.2.1 樣品前處理
稱取5 g橄欖油至燒杯中,加入20 mL正己烷,充分攪拌混勻后倒入250 mL分液漏斗中,5 mL正己烷清洗燒杯后并入分液漏斗,在分液漏斗加入30 mL正己烷飽和乙腈、10 mL飽和氯化鈉,振蕩靜置分層,棄去下層氯化鈉水溶液,收集中間乙腈層經漏斗濾入雞心瓶中(漏斗上預置無水Na2SO4層),再用30 mL正己烷飽和乙腈萃取一次,合并萃取液至雞心瓶中,40 ℃減壓旋轉蒸發至約2~5 mL,過Envi-18和PSA串聯雙柱(10 mL乙腈預活化),5 mL乙腈清洗雞心瓶(配合超聲波)后一并上柱,用乙腈繼續洗脫,全部收集,至洗脫液達到20 mL時停止,在40 ℃減壓旋轉蒸發至近干,用丙酮+正己烷(1+1)溶劑定容至1 mL上機。
1.2.2 色譜和質譜條件
氣相色譜條件:ECD,載氣為氮氣(純度為99.999%),流速2.0 mL/min,進樣口溫度為260 ℃,進樣量為1 μL,不分流進樣,檢測器溫度為300 ℃。FPD,載氣為氮氣(純度為99.999%),流速為2.0 mL/min,尾吹流量為60 mL/min,氫氣(純度為99.999%)流量為75 mL/min,空氣流量為100 mL/min。進樣口溫度為220 ℃,進樣量為1μL,不分流進樣,檢測器溫度為250 ℃。
氣相色譜-質譜條件:載氣為氦氣(純為度99.999%),恒流模式,流速為1.0 mL/min,進樣口溫度為260 ℃,進樣量為1 μL,不分流進樣,色譜-質譜接口溫度為280 ℃,離子源溫度為230 ℃,四極桿溫度為150 ℃,離子化方式為EI,電子能量為70 eV,倍增器電壓在自動調諧后加400 V。313種農藥的色譜分離譜圖見圖1, 其他相關參數見表1。
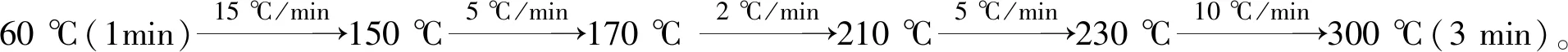
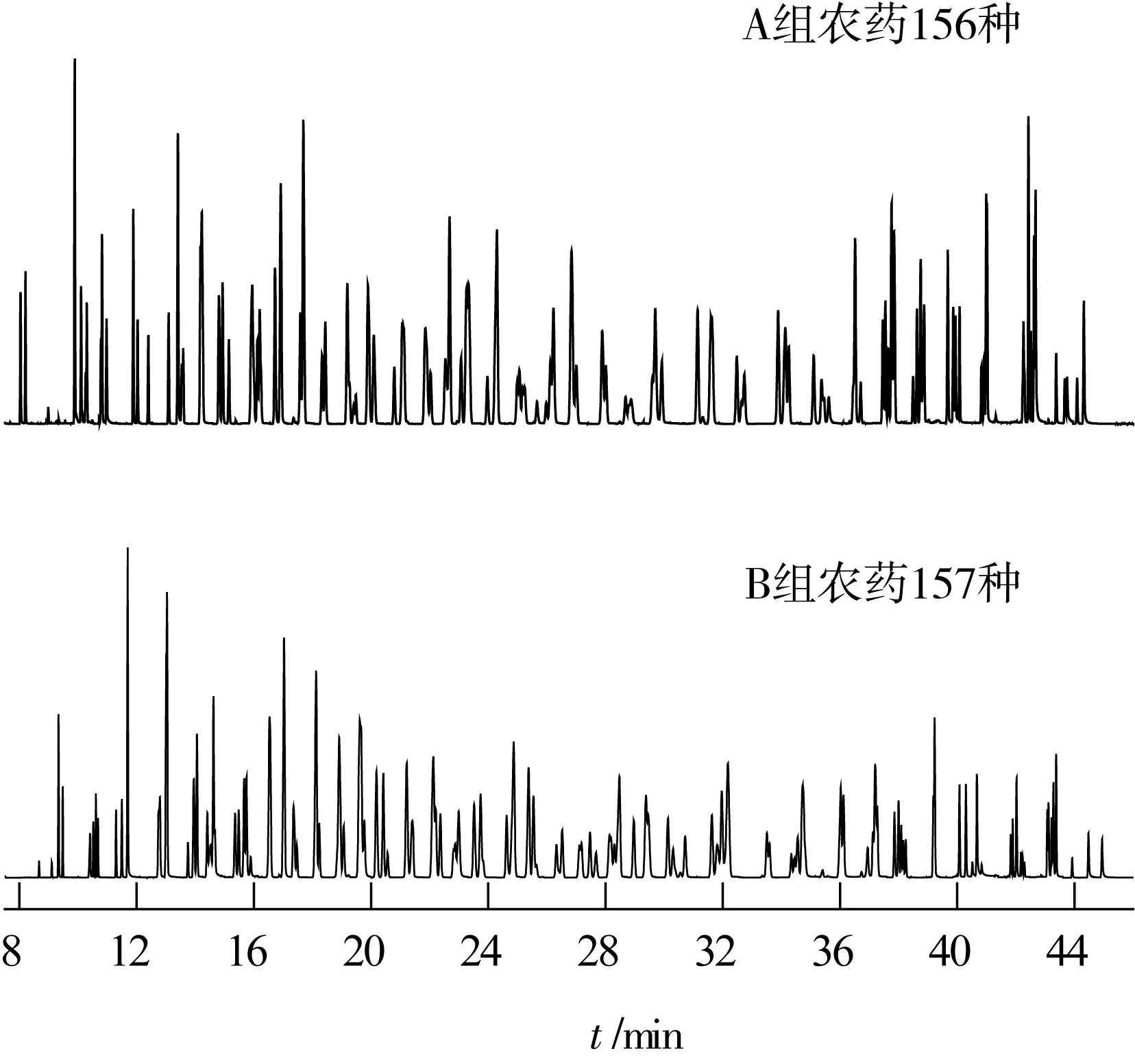
圖1 313種農藥混標選擇監測總離子流色譜圖

表1 選擇離子監測待測農藥的保留時間、定量離子、定性離子
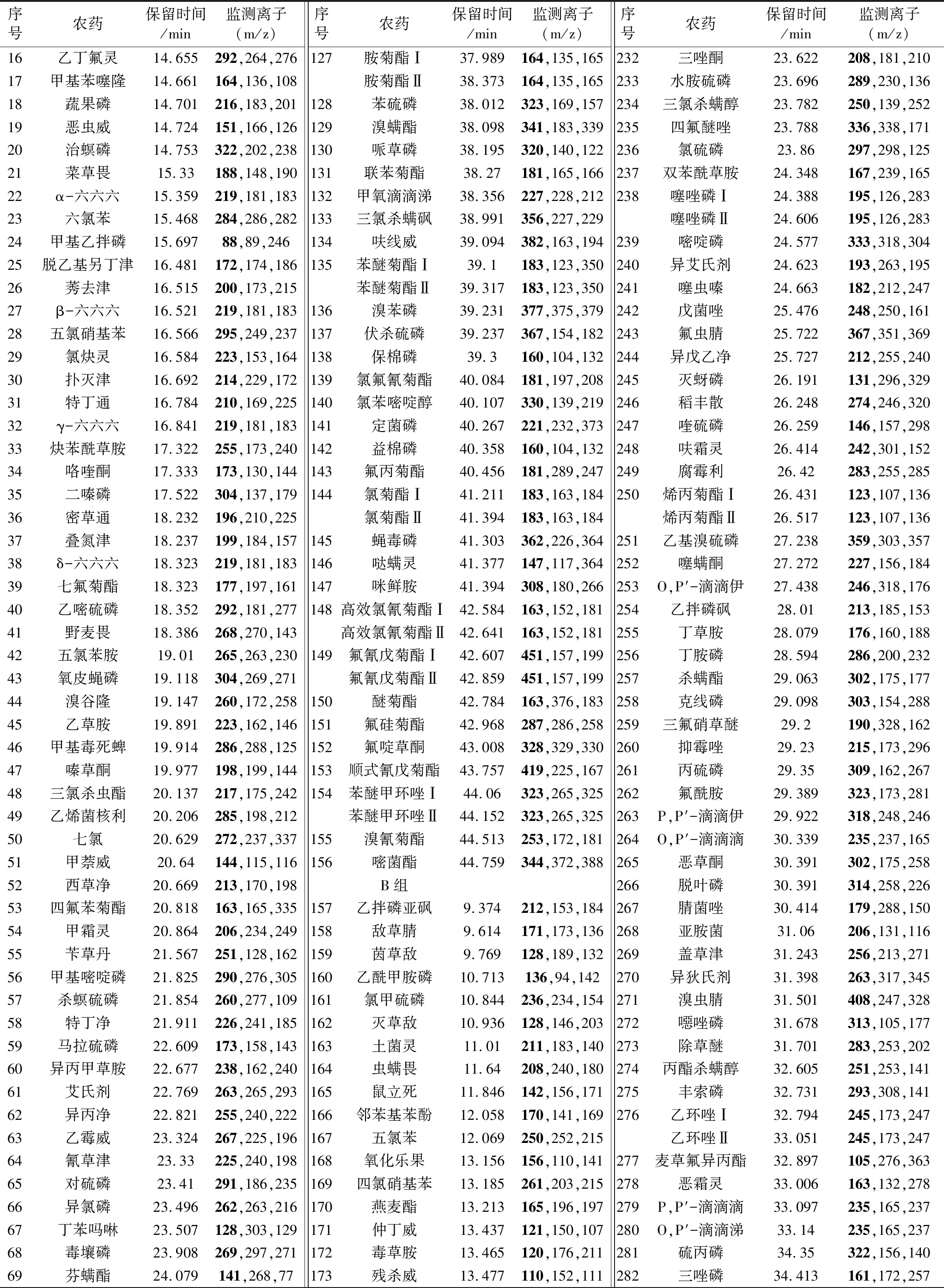
續表1

續表1
注:黑體為定量離子。
2 結果與討論
2.1 樣品前處理條件的選擇
橄欖油的主要成分為油脂,它的存在會對后續分析帶來強烈的干擾,乙腈中可溶入的油脂量極少,對絕大多數農藥溶解度則較大,故采用乙腈來作為橄欖油中農藥殘留的提取溶劑。實驗中先使用正己烷對橄欖油進行溶解,乙腈反提取,正己烷-乙腈-鹽水三相分配,其中油脂溶于正己烷相,水溶性雜質溶于水相,農藥組分轉移至乙腈相中,隨農藥組分進入乙腈相的部分油脂和其他雜質則采用SPE手段進行凈化。
試驗Envi-18柱、PSA柱、NH2柱、Carb柱、Florisil柱的各種組合對本實驗提取液的凈化效果。由于油脂是主要干擾雜質,故Envi-18柱必選;Florisil柱中的鎂化硅膠會對極性農藥產生強吸附,象甲胺磷、氧化樂果等農藥回收率極差,加強洗脫溶劑的極性也不能有效改善,故該柱由于其適用范圍窄而排除;Carb柱中含層狀石墨碳結構,某些農藥分子中含平面環狀結構,此結構與石墨碳中的六方層狀結構產生強吸附,很難洗脫,導致回收率大幅下降,若一定要洗脫來提高回收率的話,必需使用含高比例甲苯或苯的溶劑,這直接導致該柱與Envi-18柱組合使用時除油脂的效果很差,也排除;PSA柱和NH2柱效果相當,由于相同規格的PSA柱比NH2柱吸附容量大,故選用PSA柱與Envi-18柱組合成串聯雙柱。雙柱分別吸附非極性和強極性雜質,互為補充,其中,Envi-18柱的硅膠骨架上的十八烷基官能團在與乙腈配合使用時對油脂的去除有十分顯著的效果,同時還可以除去其他一些非極性的雜質如色素中的非極性組分等[5,9]。PSA柱的硅膠表面鍵合有極性官能團,它能利用氫鍵吸附的原理從樣品中吸附極性化合物[9],與乙腈配合使用時,絕大部分農藥均可定量洗脫,而強極性雜質如有機酸、部分色素等還吸附在PSA填料上。實驗時雙柱串聯,僅使用一種溶劑過柱,簡單快速,實際操作時,應在活化時先控制好流速,可采用預調節方法來實現平穩的流速:先用乙腈調節,若流出不暢,可摘下Envi-18柱讓PSA柱上的乙腈自行往下流,流速合適時迅速插上Envi-18柱,即可獲得整體平穩的流速,然后將提取液上柱,以免串聯柱內形成內壓導致洗脫液流出不暢。
2.2 氣相色譜-質譜條件的優化
一般建立氣質聯用測定幾百種農藥多殘留的儀器條件較為耗時繁瑣,本研究在配合質譜庫和相關文獻[4,7]參數的情況下,采用速成法進行:
先配置1~5 mg/L混合標準溶液,采用質譜全掃描方式、通用型氣相柱溫程序進行走樣,設定較長的高溫程序段,以確保高沸點農藥組分可以流出色譜柱,此時觀察譜圖中色譜峰的分布,對柱溫程序進行精細調節:色譜峰較密集的區域,減緩該區域對應保留時間段的升溫速率,反之,則加快速率,綜合考慮效率對柱溫進行調整,使整張譜圖中色譜峰的分布相對均勻,值得注意的是,柱溫程序的變化對出峰時間的影響具有滯后性,部分目標區域的調節效果不佳,此時將調節區域提前,可獲得不同的分離效果。
依據質譜庫和相關文獻參數確定各農藥組分的可能特征離子,再利用各農藥組分的特征離子確定保留時間,對于無法確認歸屬的少量農藥組分(如:響應值較低、同分異構體、相似結構等)則需使用單標進行確認。同時,通過調整氣化溫度對特殊農藥是否分解、是否無法氣化等加以確認,將個別無法使用氣質聯用測試的農藥剔除出去。
通過綜合考查特異性、豐度比、質量數的因素,挑選3個離子進行監測,再依據步驟2中確認的保留時間來分組,調整駐留時間(Dwell time)使每組的循環掃描次數在每秒3次左右,保證每個完整的離子流色譜峰包含足夠的采點數,以免峰形失真影響精密度。
2.3 氣相色譜-質譜測定結果
采用本法處理的某進口初榨橄欖油樣品選擇監測總離子流色譜圖見圖2(B組),如圖所示:總離子流色譜圖中沒有出現成片的或比例非常懸殊的雜質峰,在保留時間為36~36.5min有一個較大的雜質峰,經優化分離條件后,僅2個農藥(氟苯嘧啶醇、戊唑醇)在該區域出峰,這2個農藥的定量離子為m/z 314、m/z250,離子質量數較大,不受該種程度的雜質峰的影響,其他位置的干擾峰都較小。經優化監測離子,本前處理方法所處理的橄欖油中各農藥的定量離子基本不受基質干擾,有少部分農藥的定性離子有干擾,由于此部分離子僅供定性用,故其干擾幅度尚可接受。該進口初榨橄欖油中檢出的2種農藥提取離子流色譜圖如圖2所示:倍硫磷檢出值為0.032 mg/kg,腐霉利檢出值為0.11 mg/kg,m/z153譜圖基線抖動,并且在靠近該峰的前部有小幅干擾,這是因為m/z153離子質量數和豐度比均較小,抗干擾性不強,容易受到干擾,在本研究中,該離子僅供定性用,不涉及準確定量,故滿足實驗要求。其他離子m/z278、m/z169、m/z285、m/z283、m/z255的譜圖均沒有干擾,信噪比很高。
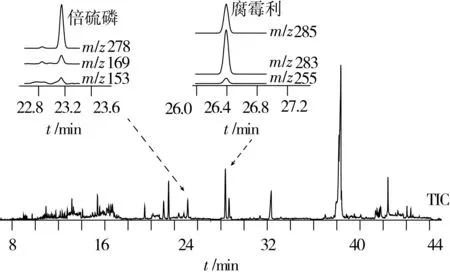
圖2 初榨橄欖油樣品選擇離子監測總離子流色譜圖及陽性農藥(倍硫磷、腐霉利)提取離子流色譜圖
2.4 氣相色譜測定結果
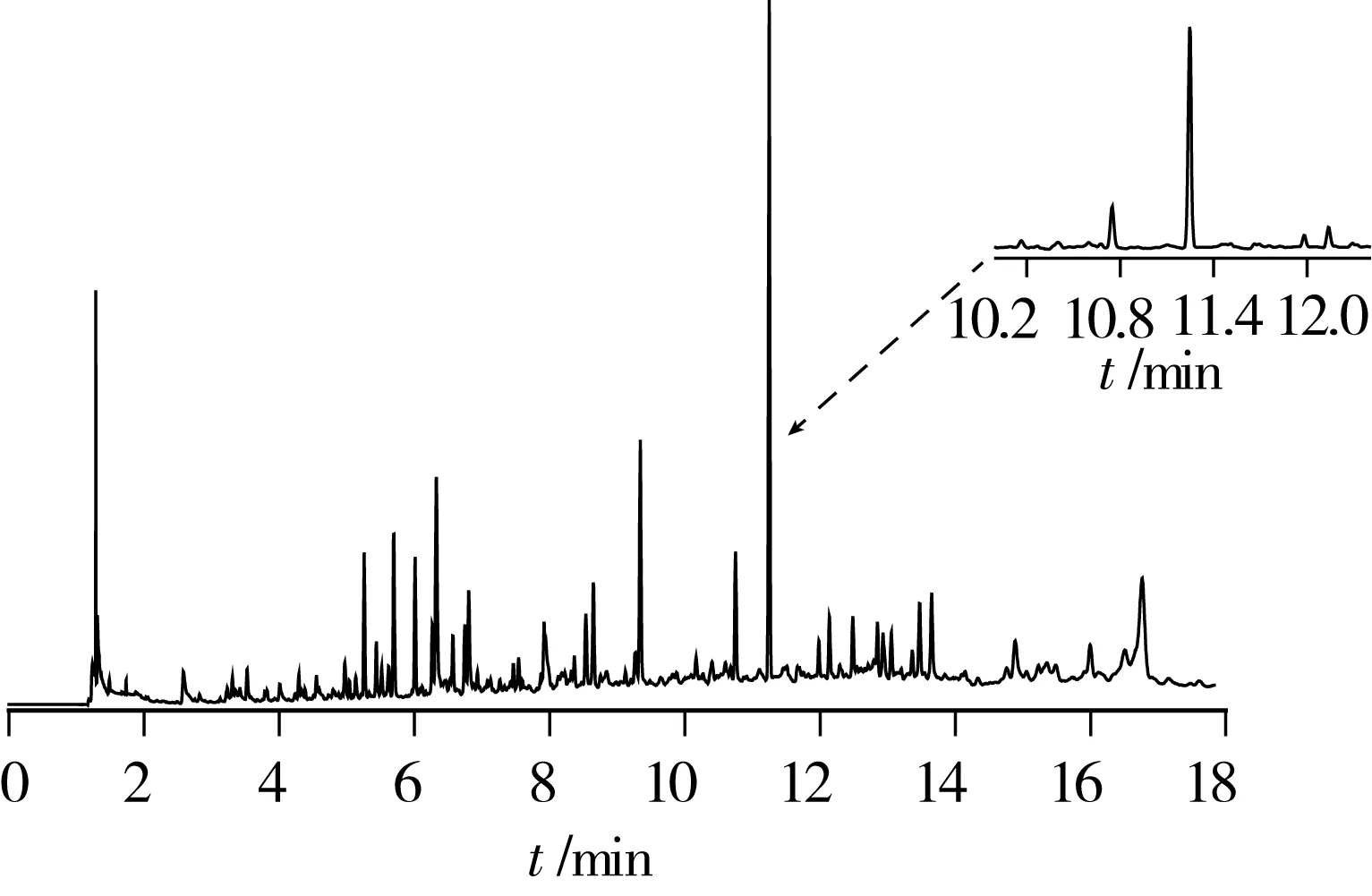
圖3 某進口初榨橄欖油樣品的GC-ECD色譜圖(檢出腐霉利)
試驗考察了本前處理的凈化方案在氣相色譜(GC-ECD、GC-FPD)上的適用性。GC-ECD譜圖見圖3:圖中有相當數量的雜質峰,這是因為ECD檢測器雖然靈敏度高,但選擇性一般,許多雜質也含有電負性基團導致在譜圖中形成干擾信號,由于干擾信號強度不大,譜圖放大后其基線情況還是可以接受的。本例進口初榨橄欖油樣品中檢出腐霉利0.11 mg/kg,其實際信噪比很高,完全滿足定量要求,由于有機氯和擬除蟲菊酯類農藥在ECD檢測器上普遍響應值很高(腐霉利算相對低的),而雜質峰的高度相對于目標物來說不算高,并且各雜質峰峰形較尖銳,所以本法中,有機氯和擬除蟲菊酯類農藥很容易通過放大譜圖、調整升溫程序或更換色譜柱實現與雜質峰的分離,進而進行定性定量。GC- FPD譜圖見圖4:由于FPD檢測器的高選擇性,未出現較大的雜質峰,在保留時間約為9.2、10.9、14.1 min處有很小的雜質峰,在保留時間約為16.8、17.7min處有輕微基線抖動。經觀察,基線抖動只是個別批次實驗時出現,大部分情況下沒有,將色譜柱設置280 ℃烘烤2 h后重新走樣,發現該處的基線回復為平線,這是由于色譜柱中殘留有少量高沸點物質,在高溫階段流出色譜柱,進入火焰光度檢測器時,影響到檢測器中氫火焰的燃燒狀態,使氫火焰的發光強度等發生變化[3],同時由于它們在氣化后體積膨脹,而色譜柱內徑較細,無法同時流出色譜柱,最終呈連續流出狀態導致基線出現輕微的抖動。由于這些雜質峰和基線抖動幅度都非常小(參照圖4中0.032 mg/kg倍硫磷的響應值),相對于有機磷農藥的響應值來說基本可忽略不計,整體信噪比非常高,可進行定性定量。對于出峰時間較早的幾種有機磷如甲胺磷、乙酰甲胺磷、氧化樂果等,GC-FPD法的檢出限比GC-MS法做得更低,均可達到0.01 mg/kg以下。由于氣相色譜上農藥的保留時間容易重疊,所以一次性測試數量較少,可依據需要配置數量合適的標液(例如十幾種一組)對需要監控的農藥進行專項測定。結合GC-MS法的后續確證,本前處理方法配合氣相色譜適用于橄欖油樣品中有機磷農藥(GC-FPD)和有機氯、擬除蟲菊酯類農藥(GC-ECD)的測定。氣相色譜測定的適用性結論僅作為本研究的附屬,故不再作進一步技術參數考察。
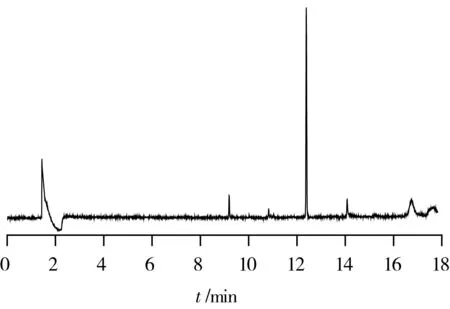
圖4 某進口初榨橄欖油樣品的GC-FPD色譜圖(檢出倍硫磷)
2.5 氣相色譜-質譜測定檢出限、線性范圍、回收率和精密度
按本方法對添加了313種農藥混標的空白初榨橄欖油樣品進行加標回收測定,以信噪比≥3確定方法檢出限,甲胺磷、三唑醇檢出限為0.03 mg/kg,乙酰甲胺磷、氧化樂果、環莠隆、苯嗪草酮、烯丙菊酯檢出限為0.02 mg/kg,其余項目的檢出限均可達到0.01 mg/kg。方法線性范圍:0.01~0.5 mg/L,相關系數R2>0.99,在樣品中添加0.01、0.05、0.20 mg/kg水平各農藥標準品,按本方法進行加標回收試驗(n=6)。三個水平回收率分布范圍見表2。
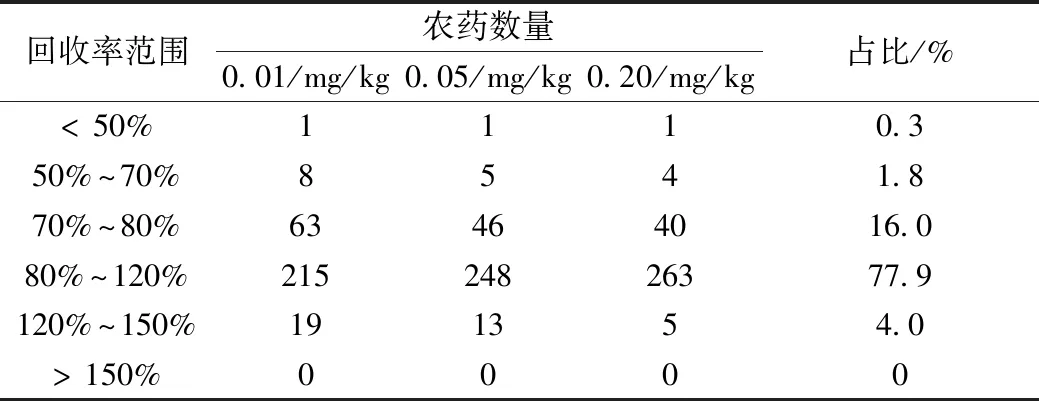
表2 橄欖油樣品在0.01、0.05、0.20 mg/kg水平加標的回收率分布范圍(n=6)
如表2所示,3個水平加標的平均回收率共313×3-7=932個數據(7個項目檢出限大于0.01 mg/kg,故其對應的0.01 mg/kg水平加標數據不參與統計),采用分步回收率監控和基質匹配標準溶液對比響應值的辦法[2]分析部分項目回收率偏差的原因:其中有一項農藥平均回收率<50%,是百菌清,經查,發現導致百菌清回收率偏低的因素有:1)由于百菌清結構式中的甲腈基團與樣品基質相互作用,導致提取效率偏低,這可以通過在提取液中加入少量磷酸來破壞這種作用力,可以提高提取率。通過使用不同樣品基質驗證該作用力的影響時,發現白蘿卜樣品基質體現最明顯:在不使用磷酸來破壞這種作用力時,乙腈對百菌清的提取率<10%或提取不出來,加入少量磷酸其提取率可達70%以上。該現象也說明對于百菌清來說,不同樣品基質的回收率參考意義不大。2)由于百菌清的易被吸附特點,對全程測試環境的潔凈度要求較高,例如常在襯管和色譜柱柱頭、柱尾等處發生吸附導致回收率偏低,若被吸附的是標液導致校準系列響應值偏低則出現回收率虛高的現象。所以一般百菌清的真實回收率能達到70%就屬于較優結果,而通用性多殘留方法中由于其前處理所用的石墨碳凈化填料對百菌清的平面芳香環結構具有強吸附,難以洗脫,回收率不超過10%,故百菌清較難并入多殘留分析方法。本實驗將百菌清并入多殘留分析方法,平均回收率44.3%,由于其精密度尚可接受(RSD<12.6%),故未將其剔除。回收率介于50%~70%的項目主要有敵敵畏、聯苯、茵草敵、五氯苯、六氯苯,由于這些農藥的蒸氣壓較低,揮發性較強,在前處理濃縮時,一部分農藥組分跟隨溶劑揮發了。回收率介于120%~150%的項目主要有氟啶脲、噁唑磷、吡唑硫磷、茚蟲威、豐索磷、噻唑磷、克螨特、氰戊菊酯、甲基對氧磷、對氧磷、噻螨酮,使用基質匹配標準溶液進行定量,農藥項目的實際回收率均在110%以內,故該部分回收率的偏離應歸因于基質增強效應[21-22]。由于目標物濃度的提高能減弱基質效應的影響,故加標水平為0.20 mg/kg的樣品中,其回收率介于120%~150%的項目數量大幅減少。總體上看,回收率在70%~120%之間的項目占絕大多數(93.9%),滿足GB/T 27404—2008 《實驗室質量控制規范 食品理化檢測》的要求,個別農藥項目回收率不甚理想但對于多殘留分析來說尚處于可接受范圍,RSD<17.8%,結果滿意。目前檢測植物油中農藥殘留項目數最多的標準是SN/T 4428—2016《出口油料和植物油中多種農藥殘留量的測定 液相色譜-質譜/質譜法》(77種農藥),該標準采用乙腈提取,分散固相萃取凈化,液相色譜-串聯質譜檢測,相對來說,本法同時測定313種農藥,速度更快,適用范圍更廣。后續試驗采用本法對大豆油、花生油、葵花籽油進行測定,發現在氣相色譜-質譜選擇監測模式下,定量離子和定性離子的譜圖和橄欖油的譜圖區別不大,說明本法的凈化方案亦適用于其他植物油的前處理凈化。
2.6 實際樣品分析
按本研究方法對34份進口初榨橄欖油進行檢測,結果顯示3份檢出農藥殘留,其中一份檢出倍硫磷0.032 mg/kg、腐霉利0.11 mg/kg。另外2份均檢出腐霉利殘留,檢出值分別為0.064、0.15 mg/kg。其余樣品中各項目均為未檢出。
3 結論
本研究基于串聯雙柱凈化方案建立了橄欖油中313種農藥多殘留檢測的前處理方法,項目覆蓋面廣,操作快速,能有效去除油脂樣品中大量干擾雜質。配合氣相色譜和氣質聯用進行測定,所得GC-MS、GC-ECD、GC-FPD譜圖均呈現良好的凈化效果。同時,采用本方法對其他植物油(大豆油、花生油、葵花籽油)進行測定,亦獲得滿意的結果。
