高碘酸鈉選擇性氧化對亞麻短纖維性能的影響
2019-07-30 12:24:58高潔,佟瀟,崔瑩,王馨,趙波
印染助劑 2019年6期
高 潔,佟 瀟,崔 瑩,王 馨,趙 波
[1.齊齊哈爾大學輕紡學院,黑龍江齊齊哈爾 161006;2.齊齊哈爾大學圖書館,黑龍江齊齊哈爾 161006;3.日華化學研發(上海)有限公司,上海 201620]
亞麻短纖維屬于天然纖維素纖維,纖維素纖維的選擇性氧化分為伯羥基氧化和仲羥基氧化。對于仲羥基氧化,Jackson 等[1]用過碘酸對淀粉和纖維素進行氧化,發現過碘酸只將兩個仲羥基氧化成醛基,而伯羥基則沒有發生變化。Silvia Vicini 等[2]用高碘酸氧化以棉和麻為主的纖維素材料,并對其力學性能進行剖析,結果表明:材料的氧化降解程度和纖維的自然老化存在一定的相關性。鄭培培等[3]用高碘酸鈉對苧麻纖維進行處理,研究了苧麻纖維的結構與性能,為進一步實現苧麻纖維的功能改性提供參考。對于伯羥基氧化,de Nooy 等[4]在磷酸溶液中以NaNO3為氧化劑,NaNO2為催化劑氧化纖維素,可使90%以上的伯羥基氧化成羧基,但是該體系必須在4 ℃下反應,否則會引起纖維較大程度的降解。盡管體系對伯羥基有極強的氧化能力,但在氧化過程中纖維素大分子降解劇烈,因此對降解機理以及抑制降解方法的研究一直是關注的焦點[5]。經過氧化處理的纖維,由于引入了新的官能團具有新功能和新用途,應用領域得到了擴寬,如可以獲得具有熒光、儲能、螯合劑及生物醫用等特殊功能的高分子材料[6-7]。本實驗采用高碘酸鈉對亞麻短纖維進行選擇性氧化,改善可紡性,使其綜合性能得到提升,以滿足紡紗技術的要求。
1 實驗
1.1 材料及儀器
亞麻短纖維(齊齊哈爾金亞亞麻紡織有限責任公司,纖維長度56 mm,斷裂強度12.05 cN/dtex,分裂度237 Nm);D8 X 射線衍射儀(德國布魯克公司)。
1.2 高碘酸鈉選擇性氧化亞麻短纖維
亞麻短纖維雜質含量偏高,可紡性能差,在進行氧化改性之前,必須進行煮漂處理。經過煮漂處理的短纖維中果膠、木質素等雜質會被部分去除,分裂度和白度均有一定程度的提高,后續高碘酸鈉氧化才能順利進行。
稱取一定質量經過前處理的亞麻短纖維置于棕色錐形瓶中,加入預先配好的高碘酸鈉溶液,避光氧化處理相應時間,得到氧化的亞麻短纖維。將氧化后的亞麻短纖維于0.1 mol/L 丙三醇溶液中浸泡45 min,用去離子水充分洗滌,然后置于空氣中晾干,放入干燥器中,待恒重后稱質量。
1.3 測試
X 射線衍射:將亞麻短纖維剪成粉末狀并安放在X 射線衍射儀的玻璃樣品架上,在穩定條件下測試,得到X 射線衍射強度曲線,再用Origin 8.1 軟件進行分峰擬合。測試條件:管電壓為40 kV,管電流為30 mA,掃描范圍為10°~40°,用Ni 濾波,Cu 靶Kα 射線,掃描速率為2°/min。
斷裂強度:參考GB 5882—86,采用YG001A 纖維電子強力儀進行測試,按下式計算斷裂強度,取5 個試樣的平均值。
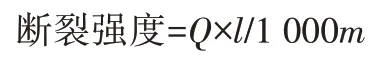
式中:Q為麻束的斷裂強力,cN;m為麻束的切割質量,mg;l為麻束的切割長度,40 mm。
分裂度:隨機抽取試樣梳理,在中間部位截取10 mm,在電子天平上稱取1 mg 左右的5 個試樣,逐根計數,按下式計算分裂度,取5個試樣的平均值。
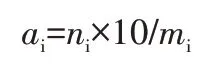
式中:ai為第i份纖維的分裂度;ni為第i份纖維根數;mi為第i份纖維質量,i=1,2,3,4,5。
2 結果與討論
2.1 影響分裂度及斷裂強度的因素
2.1.1 高碘酸鈉用量
從表1 中可以看出,隨著氧化劑NaIO4用量的增加,斷裂強度不斷下降。主要原因是亞麻短纖維在氧化過程中,隨著氧化劑NaIO4向短纖維內部不斷擴散與滲透,纖維素葡萄糖環上的C2和C3鍵打開,C2和C3位置的羥基氧化成醛基[8],晶區部分被破壞,使亞麻短纖維分子上的羥基減少,結晶度降低,分子間作用力減小,從而使超分子結構存在缺口、弱點。在拉伸時弱點首先斷裂,缺口逐漸擴大,進而應力集中,分子鏈拉斷,導致纖維斷裂強度降低。
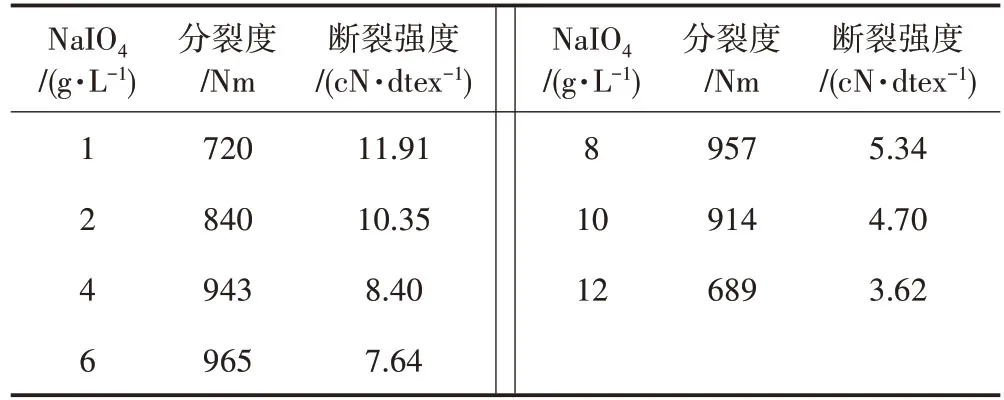
表1 高碘酸鈉用量對分裂度及斷裂強度的影響
隨著NaIO4用量的增加,分裂度呈現先增大后減小的趨勢,當氧化劑用量為4~8 g/L 時,分裂度變化比較平緩;當用量大于8 g/L 時,分裂度則不斷下降。這是因為亞麻短纖維經過煮漂前處理后,已由束纖維向單纖維轉變,在氧化過程中纖維被潤濕,膨潤程度增大,分子間的氫鍵部分也受到了破壞,纖維變細,分裂度逐漸增大并保持一定值;而氧化劑用量大于8 g/L 時,亞麻短纖維幾乎完全呈現單纖維狀態,纖維短絨率增加,短纖維損傷增大,分裂度降低。
綜合考慮分裂度和斷裂強度,高碘酸鈉的最佳用量為6 g/L。
2.1.2 反應時間
由表2 可知,隨著反應時間的延長,斷裂強度先急劇下降再緩慢下降。這是由于反應初期,氧化劑只作用在纖維的無定形區,由于分子結構疏松,分子間作用力小,氧化劑與纖維素葡萄糖環上羥基的反應更易于發生,因此,斷裂強度先急劇下降。但反應50 min 后,氧化劑NaIO4開始逐漸進入短纖維的結晶區,結晶區分子結構相對緊密,反應速率減慢。因此斷裂強度下降也變緩慢。

表2 反應時間對分裂度及斷裂強度的影響
隨著反應時間的延長,分裂度呈現先增大后減小的趨勢。這是由于隨著反應時間的延長,短纖維內部結構逐漸疏松,束纖維向單纖維轉變,使分裂度逐漸增大;當反應時間超過60 min 后,因為長時間的氧化作用使纖維分裂過度,短絨率增加,致使分裂度下降。因此,要注意控制反應時間,使短纖維的分裂程度相對適中,保證其可紡性。
從斷裂強度和分裂度的角度綜合考慮,反應時間控制在50~60 min為宜。
2.1.3 反應溫度
從表3 可以看出,隨著反應溫度的升高,亞麻短纖維的斷裂強度逐漸降低。這是因為溫度升高使氧化劑分子和纖維素分子的熱運動加快,氧化反應速率加快,同時也使短纖維的水解速率增大,導致短纖維分子中的葡萄糖環和甙鍵斷裂的可能性增大。短纖維無定形區的價鍵結構相對較弱,氧化反應首先發生在無定形區,隨著氧化條件的變化,氧化劑逐漸向結晶區和內部滲透,破壞了纖維分子鏈間的氫鍵、范德華力等次價鍵結構,同時伴隨著纖維的降解,因此短纖維的結晶度降低,斷裂強度也降低。
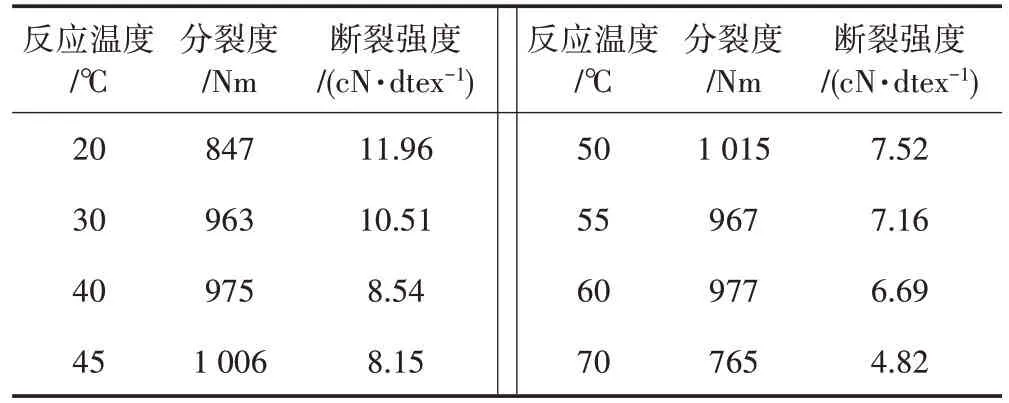
表3 反應溫度對分裂度及斷裂強度的影響
反應溫度較低時,分裂度隨著溫度的升高而增大,而反應溫度為45~55 ℃時,分裂度變化比較平緩,隨后分裂度逐漸下降。主要原因是反應初期溫度相對較低,反應速率較慢,短纖維的分裂程度也不足,因此分裂度提高緩慢。反應溫度較高時,纖維的氧化裂解程度增大,短絨率增加較快,分裂度反而降低。
綜合考慮斷裂強度和分裂度,反應溫度控制在45~50 ℃為宜。
2.2 X 射線衍射
從圖1 中可以看出,氧化前后亞麻短纖維的特征吸收峰峰形相似,呈現出典型的結晶纖維素Ⅰ的X射線衍射曲線,2θ=14.77°、16.69°、22.77°對應纖維素的101、、002 晶面的衍射峰,這是典型的纖維素Ⅳ結構[9]。其中一個主要峰值是002 晶面峰所代表的晶體結晶區衍射強度;次要峰值是無定形區峰所代表的峰值強度,即和002晶面之間波谷的位置。
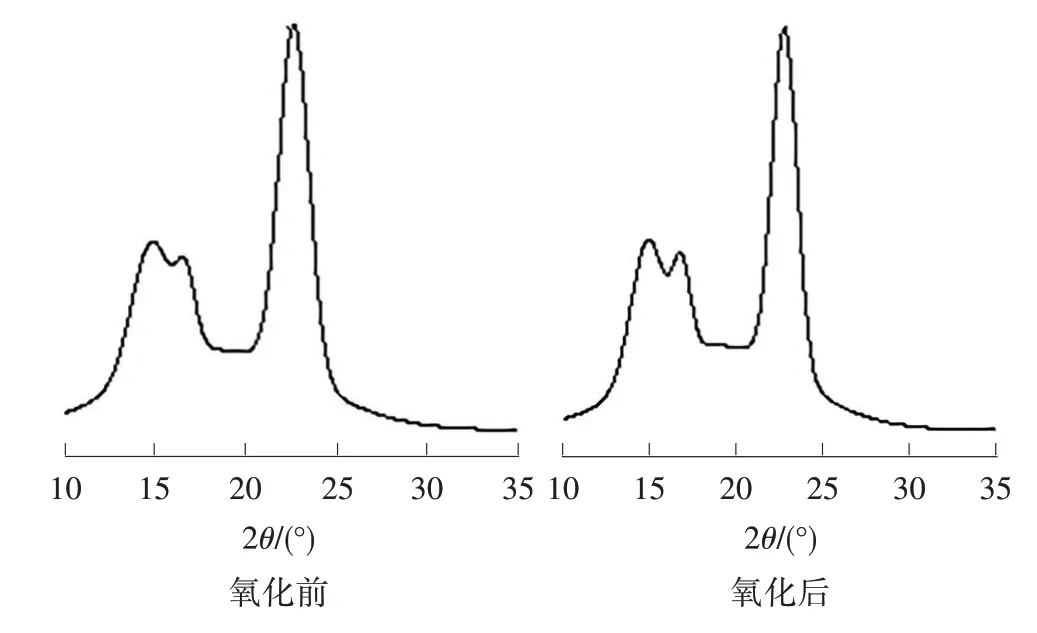
圖1 亞麻纖維氧化前后的XRD 譜圖
氧化處理后的亞麻短纖維2θ衍射角并沒有發生太大的變化,均在14.77°、16.69°和 22.77°左右,這說明氧化處理后的亞麻短纖維纖維素結構在本質上并沒有發生變化。但是氧化后的亞麻短纖維衍射峰強度比原亞麻短纖維低一些,說明纖維素纖維的結晶區部分受到了破壞。氧化劑與纖維素纖維反應首先作用在纖維的無定形區,隨著反應程度的加深,氧化劑會作用在纖維的結晶區,引起結晶度下降。
3 結論
用高碘酸鈉對亞麻短纖維進行氧化,最優處理工藝條件為:氧化劑高碘酸鈉用量6 g/L、45~50 ℃反應50~60 min;氧化后亞麻短纖維的綜合性能得到較大改善,分裂度為965 Nm,斷裂強度為7.64 cN/dtex,結晶度下降,滿足紡紗加工技術要求。
