脾臟炎癥性肌纖維母細胞瘤一例
2019-09-03 08:46:50王守乾管清春楊陽唐朝輝王英超
肝膽胰外科雜志 2019年8期
王守乾,管清春,楊陽,唐朝輝,王英超
(吉林大學第一醫院 肝膽胰外二科,吉林 長春 130000)
病例
患者男性,70歲,既往體健,此次因“間斷性腹脹20天”入院。查體:未見陽性體征。實驗室檢驗:血常規:紅細胞4.13×1012/L,血紅蛋白91 g/L,血小板352×109/L。腫瘤標志物及其他實驗室檢驗均正常。輔助檢查:腹部、盆腔多排CT平掃+三期增強,提示脾臟見團塊狀異常,其內密度不均,CT值約26~44 Hu,中心見多發結節樣鈣化影,三期增強掃描呈明顯不均勻強化(見圖1)。排除手術禁忌后行腹腔鏡脾切除術,術后病理結果:脾臟炎癥性肌纖維母細胞瘤,伴EBV相關性淋巴組織增生性病變,間質伴大量玻璃樣變性、肉芽腫及多核巨細胞(見圖2)。
討論
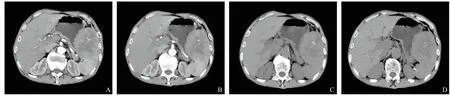
圖1 腹部、盆腔多排CT平掃+三期增強提示明顯不均勻強化,脾臟內見結節樣鈣化影
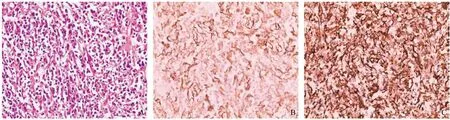
圖2 病理檢查(HE,×400)及免疫組化(×400)
炎癥性肌纖維母細胞瘤(imflammatory myofibroblastic tumor,IMT),是由分化的肌纖維母細胞性梭形細胞組成的,常伴大量漿細胞和(或)淋巴細胞的一種腫瘤[1]。曾命名為漿細胞肉芽腫、炎性假瘤、纖維黃色肉芽腫、炎性肌纖維組織細胞增生、黏液樣錯構瘤等。目前其病因尚不清楚,但已知與IMT相關的一種病原體是Epstein-Barr病毒(EBV)[2],本例亦合并EBV感染。該病主要見于兒童和青壯年,肺為最常見的發病部位,也可發生于肺外其他器官[3],如胰腺[4]、闌尾[5]、心臟[6]等其他部位,而發生于脾臟炎癥性肌纖維母細胞瘤(spleen imflammatory myofibroblastic tumor,SIMT)十分罕見。
SIMT在癥狀上可表現為左上腹或上腹痛,發熱、乏力、體重下降、貧血等,當SIMT較大時,周圍臟器可能因受壓而出現相應癥狀,本例則以間斷性腹脹為首發癥狀。SIMT多無明顯常見體征。影像學方面,SIMT在MRI T1加權圖像上通常是等或低信號,但T2信號可有變化[7]。有報道稱用“釓”這種試劑進行MRI檢查時可導致SIMT相對于正常脾組織的信號強度增加[8-9]。SIMT在增強CT上可表現為三期不均勻強化,伴或不伴有鈣化[9-10],本例三期增強掃描呈明顯不均勻強化,且中心見多發結節樣鈣化影,符合文獻報道。在超聲檢查中,SIMT表現為異質性,低或高回聲腫塊[11-12]。實驗室檢查可有多克隆性高球蛋白升高、ESR升高、血鈣升高、血小板增多、紅細胞和血紅蛋白下降等[7]。本例與文獻報道一致。本例術前未曾考慮SIMT,故未行多克隆性高球蛋白及ESR檢驗。值得一提的是,SIMT在癥狀、體征、影像學及實驗室檢查方面均無特異性表現。
SIMT的診斷最終依賴于組織病理學及免疫組學。SIMT在鏡下主要以肌成纖維細胞和成纖維細胞等梭形細胞增生為主[13],常伴有大量混合性慢性炎細胞浸潤。這些慢性炎細胞以淋巴細胞、漿細胞、巨噬細胞、多核巨細胞為主[14-15],梭形細胞間可有不同程度的纖維化、玻璃樣變性、鈣化或壞死,本病例符合SIMT以上病理組織學特點。SIMT在免疫組化方面主要表現為Vimentin(波形蛋白)陽性、SMA(平滑肌肌動蛋白)陽性,本例Vimentin及SMA均陽性,符合SIMT免疫組化特點。SIMT主要應當與以下疾病相鑒別:(1)炎性假瘤樣濾泡細胞腫瘤[16-17](in flammatory pseudo tumor,IPT):其與SIMT的組織病理學及免疫組化表型特點極為相似,但IPT在免疫組化方面CD21及CD35標記多呈陽性,借此可進行鑒別。(2)脾臟漿細胞瘤[18]:腫瘤細胞呈致密、彌漫分布,漿細胞不成熟,核異型,可見核分裂象,盧氏小體極少或缺如。漿細胞肉芽腫型SIMT則為成熟漿細胞,盧氏小體易見。(3)惡性纖維組織細胞瘤[19]:腫瘤細胞成分混雜與SIMT相似,但前者含異型細胞,核分裂易見,后者無細胞異型及核分裂象,必要時可行細針穿刺細胞學檢查。(4)惡性淋巴瘤[20]:本病多伴淋巴結和脾臟腫大,腫瘤多由單一淋巴細胞組成,少有混合性、慢性炎細胞浸潤。結合病史、組織病理學、免疫組化多可進行鑒別。此外SIMT還應當與其他原發和繼發的脾臟占位性病變如脾血管瘤、脾轉移瘤、脾錯構瘤等進行鑒別。
SIMT的治療以手術切除為主,且預后多良好。SIMT的自然轉歸目前尚未明確,但有報道稱SIMT手術切除后有復發和轉移,尤其以肝轉移多見[21],故術后需密切隨訪。
總之,SIMT是發生于脾臟的一種罕見病,臨床表現上無特異性,其診斷依賴于術后組織病理學及免疫組織化學檢查,治療以手術切除為主,術后需密切隨訪。
