CRISPR/Cas9介導煙草多基因編輯體系的應用
2019-09-04 12:20:04謝小東高軍平李澤鋒張劍鋒魏攀羅朝鵬王晨武明珠翟妞楊軍
中國煙草學報 2019年4期
關鍵詞:煙草
謝小東,高軍平,李澤鋒,張劍鋒,魏攀,羅朝鵬,王晨,武明珠,翟妞,楊軍
1 中國煙草總公司鄭州煙草研究院,國家煙草基因研究中心,鄭州高新技術產業開發區楓楊街2號 450001;
2 湖南中煙工業有限責任公司技術研發中心,長沙市勞動中路386號 410008
基因編輯技術可以精準地對基因組中的靶位點進行缺失、敲入以及核苷酸修正等操作。CRISPR/Cas9(clustered regularly interspaced short palindromic repeats/CRISPR-associated nuclease 9,Cas9)基因組編輯技術問世以來,彌補了第一代的鋅指核酸酶(zinc finger nucleases,ZFN)和第二代的轉錄激活效應子核酸酶(transcription activator-like effector nucleases,TALEN)基因編輯技術的諸多不足。因CRISPR/Cas9系統靶向編輯基因的高效性、特異性和設計的便捷性,已成為當前最主流、最熱門的基因組編輯技術,在大量物種中得到了廣泛應用。
CRISPR/Cas9是細菌和古細菌在長期演化過程中形成的一種適應性免疫防御系統,可抵抗病毒或外源性DNA的侵染。CRISPR/Cas9編輯技術是利用導向RNA識別靶位點,并引導Cas9蛋白切割靶序列[1-2]。CRISPR/Cas系統有3種類型(Type Ⅰ、Type Ⅱ和Type Ⅲ)。現在廣泛用于基因編輯技術的CRISPR/Cas系統是根據Type II型系統改造而來的。Type Ⅱ型系統由Cas9核酸酶、pre-crRNA和反式激活crRNA組成。在前導序列的調控下,CRISPR的間隔序列被轉錄成為pre-crRNA。借助tracrRNA,RNase III再將pre-crRNA加工為成熟的crRNA,并形成具有切割活性的tracrRNA-crRNA-Cas9復合體。隨著對CRISPR/Cas9技術的不斷優化,crRNA和tracrRNA被融合成為一條單鏈引導RNA(singleguide RNA,sgRNA)。sgRNA的表達通常用III型RNA聚合酶啟動子轉錄,PAM(protospacer adjacent motif)序列決定CRISPR/Cas9系統切割的位點[3]。隨著基因編輯技術的迅速發展,科學家開發了不同CRISPR/Cas9系統,不斷在提升CRISPR/Cas9基因編輯技術應用價值。
2013年,科學家首次利用CRISPR/Cas9系統實現了對擬南芥[4]、水稻和小麥基因組的定點編輯[5],證明了該技術在植物生物學研究領域具有極高的應用價值。隨后CRISPR/Cas9系統被廣泛應用于高粱[6]、玉米[7]、甜橙[8]、番茄[9,10]、大豆[11]、大麥[12]、甘藍[12]、棉花[13]、甘蔗[14]等作物中,在性狀改良、基因調控、抗性育種以及高通量突變體文庫創制等方面得到了廣泛應用[15]。高軍平等利用CRISPR/Cas9系統分別對煙草NtPDS和NtPDR6基因實現了定點突變[16],首次在普通煙草中建立了CRISPR/Cas9技術體系。本課題組利用CRISPR/Cas9技術在煙草中鑒定了一個腋芽生長相關基因NtPIN4,將基因突變后煙草腋芽數目比野生型明顯增多,且突變類型和表型均能夠穩定遺傳至下一代[17]。潘洪杏等利用CRISPR/Cas9技術對煙草馬鈴薯Y病毒(Potato Y virus,PVY)基因eIF4E進行了定向編輯[18]。姚恒等利用CRISPR/Cas9技術對煙草茉莉酸信號關鍵轉錄因子基因NtabMYC2進行了編輯,獲得了不同敲除類型的突變體[19]。
隨著基因編輯技術的快速發展,在許多物種中同時編輯多個基因的技術逐步成熟。在楊樹毛白楊中,通過構建多靶點編輯載體系統,實現了同時敲除楊樹基因組上兩個同源的PDS編碼基因(PtPDS1和PtPDS2)[20]。在水稻和擬南芥中,構建的多基因敲除體系能夠成功突變擬南芥和水稻基因家族的多個基因、以及單個基因的多個靶點,在轉化體第一代就獲得有表型的突變體[21]。利用CRISPR/Cas9系統在水稻育種中間材料中同時編輯TMS5、Pi21和Xa13基因,成功將具有優良性狀的育種中間材料快速開發成優質多抗的兩系不育系[22]。通過構建多基因敲除體系,同時對編碼擬南芥核糖體大亞基AtRPL10亞家族的三個同源基因AtRPL10A,AtRPL10B及AtRPL10C進行了編輯,轉基因植株的三個同源基因均存在基因突變[23]。
煙草是異源四倍體農作物,多基因編輯體系的建立與應用,對加快煙草基因功能的研究和遺傳改良的有著重要的意義。在煙草中,針對多個基因、尤其是不同性狀相關基因的同時編輯的研究尚未有報道。為此,基于煙草中已有的CRISPR/Cas9系統,本文選取以下5個靶基因開展CRISPR/Cas9介導多基因編輯研究。eIF4E是轉錄起始因子(eukaryotic translation initiation factors 4E),PVY病毒通過自身的基因組連接蛋白(viral genome-linked protein,VPg)與植物的特定eIF4E基因家族成員相互作用,從而啟動病毒在植物體內的翻譯與增殖。eIF4E基因突變會導致其表達的蛋白結構改變或缺失,使病毒無法利用eIF4E蛋白侵入植株[24]。本生煙、辣椒以及普通煙草中突變或者下調eIF4E基因,均能使植株對PVY產生一定的抗性[25,26]。TOM1和TOM3是煙草花葉病毒(Tobacco mosaic virus,TMV)的抗性相關基因,研究發現TOM3蛋白與TMV病毒特異的結合,進而將病毒復制復合體整合到質膜之上,保證病毒的復制[27]。Sunil Kumar等通過構建TOM1和TOM3的干擾載體,進而下調煙草中的這兩個基因,發現轉基因植株對TMV的抗性明顯增強[28]。在本氏煙中克隆了擬南芥同源基因NbTOM1,分析證明病毒誘導NbTOM1沉默后,幾乎完全遏制了本氏煙中TMV的復制,同時干擾沙姆遜煙草的TOM1和TOM3基因后,發現轉基因煙草能夠有效地抑制TMV的生長和擴散[29]。課題組前期克隆了煙草植物螯合肽基因NtPCS1,利用RNAi技術降低NtPCS1基因的表達量之后,煙草葉片中的鎘含量顯著降低。利用TILLING技術將鎘轉運基因NtHMA2的突變后,在鎘脅迫下,與對照相比突變體煙葉中的鎘含量降低36.84%[30]。本文通過對上述5個煙草不同性狀的基因開展CRISPR/Cas9多基因編輯的探索研究,旨在為煙草多個(性狀)基因的功能研究和遺傳改良提供技術依據。
1 材料與方法:
1.1 材料
普通栽培煙草K326無菌苗培養于國家煙草基因研究中心的人工氣候室:將K326種子放入1.5 mL離心管中,加入1 mL 15% 次氯酸鈉溶液消毒15 min,無菌水清洗5~6次,用牙簽播種于MS固體培養基中。MS固體培養基成分為:4.4 g/L MS(Murashinge & Skoog Basal Mediun) 培養基、30 g/L蔗糖、2.5 g/L植物凝膠。培養條件為相對濕度60%,光照16 h/28 ℃,黑暗8 h/25 ℃。
1.2 方法
1.2.1 sgRNA設計及載體構建
利用煙草基因組序列信息,通過在線工具CRISPR MultiTargeter(http://www.multicrispr.net/index.html),設計了如下的編輯位點(表1)。將各靶位點序列分別接上U26啟動子和終止后依次串聯,并在串聯的5'端和3'引入酶切Bsa I位點后合成序列,然后利用BsaI酶切,并與預先用BsaI酶切過的CRISPR/Cas9載體連接。連接體系:酶切載體 3 μL,退火產物 2 μL,Solution I 5 μL。16 ℃連接30 min,取連接產物轉化 DH5α感受態細胞,37 ℃培養 12~16 h。用 U26-JC F:5'-TTAGGTTTACCCGCCAATA-3'和各靶序列下游引物對菌斑進行陽性克隆的PCR擴增、克隆和測序篩選。
1.2.2 煙草PCS1、eIF4E、TOM3、TOM1和HMA2基因敲除靶位點的cDNA克隆
以中國煙草基因組數據庫中PCS1、eIF4E、TOM3、TOM1和HMA2基因的cDNA序列,設計可擴增PCS1、eIF4E、TOM3、TOM1和HMA2sgRNA序列的cDNA片段引物。PCS1-cl F:5'-GAAGACTCCAAGACTACCGGG-3',PCS1-cl R:5'-GCCAATTCGCAACTAACTTCA-3',eIF4E-cl F:5'-GTAGTCGACGATGGACCTGAA-3',eIF4E-cl R:5'-CATGAAATATGAAGCCAATCG-3',TOM3-cl F:5'-GGATGGGCTTAGACCTAGTTT-3',TOM3-cl R:5'-TTAGCGAATAGGGTGGTACTG-3',TOM1-cl F:5'-TGTTGTAAATGGAGTTCGTGC-3',TOM1-cl R:5'-CTCTTTTTGGAGGCAATTTTC-3',HAM-cl F:5'-GCATTAGCAACAGCTGACATT-3',HAM-cl R:GCGATGAGAATGAAAACCAC-3',以 K326品種的cDNA為模板進行PCR擴增反應。PCR反應體系(25 μL):基因上、下游引物(10 μM)各1 μL、DNA樣品(100 ng/μL)1 μL、Taq酶12.5 μL,加 ddH2O補足至 25 μL。PCR 反應程序:94 ℃ 預變性5 min;94 ℃變性 30 s,58 ℃退火30 s,72 ℃ 延伸40 s,30個循環;72℃延伸10 min,4℃保溫。待PCR結束后進行瓊脂糖凝膠電泳,回收純化PCR產物。用TOPO-TA PCR克隆試劑盒進行TA克隆,轉化DH5α感受態細胞,37 ℃培養12~16 h,菌落PCR陽性單克隆送測序。
1.2.3 敲除載體轉化農桿菌及煙草遺傳轉化
將測序成功的單菌落擴大培養,提取質粒,利用電轉法將重組編輯載體轉入農桿菌GV3101。具體的遺傳轉化方法見王姍姍 等[33]。
1.2.4 遺傳轉化株的獲得及突變位點檢測
待再生植株的根系生長良好后,移出組培瓶于基質中,置于培養條件為相對濕度60%,光照16 h/28 ℃、黑暗8 h/25 ℃人工氣候培養箱中培養。采集適量煙株葉片,提取基因組DNA。具體步驟參照GeneAnswer GenePure新型植物基因組DNA快速提取試劑盒說明書。設計Cas9蛋白基因檢測引物,Cas9-JC F:5'-CTCAACACAACATATACAAAACAAA-3',Cas9-JC R 5'-CTTTGGCCATCTCGTTTGA-3',以各植株DNA為模板進行PCR擴增,確認外源DNA片段是否已經插入到植物基因組中。PCR反應體系和程序同1.2.2。
將滾子從動件的滾子中心視為尖底從動件的尖底,則滾子從動件的凸輪機構即成為尖底從動件的凸輪機構,因此文中僅研究后者。
設計擴增包含靶位點的150bp-180bp DNA片段的引物,以檢測目的基因是否成功被編輯。PCS1-JC F:5'-ATTGCCAGCAGTAACTTTGATGTT-3',PCS1-JC R:5'-GCCTAGTCCTAATTTTTTACTTGCC-3',eIF4EJC F:5'-ATTGGACAATGAGCTTTAGTAAGGG-3',eIF4E-JC R:5'-TTCATTTGCAGCATTCTTGGTC-3',TOM3-JC F:5'-ATGAAAGGTGGAGTAAT ACTTGTCT-3',TOM3-JC R:5'-TCAATCAT ATCCAGGTAAGAAAAGT-3',TOM1-JC F:5'-CTGTTCATTGGGAATTCGTAT TOM1-JC R:5'-GGGAGGAGAATGTAGTGAGGAA,HMA2-JC F:5'-AGCAGGTTATCCATTGGTTTG-3',HMA2-JC R:5'-TGACATCTTGGAACACACACC-3'。分別在以上各正向引物的5'端前加ggagtgagtacggtgtgc,反向引物5'前加gagttggatgctggatgg。以Cas9陽性煙株DNA為模板,進行PCR擴增。PCR反應體系和程序同1.2.2。待PCR結束后進行瓊脂糖凝膠電泳,備用進行第二輪擴增。第二輪PCR 反應體系(20 μL):ddH2O 9.0 μl、Hi-TOM Mix 10 μl、第一輪 PCR 產物1.0 μl。94℃,預變性 2 min;94 ℃變性 30 s,58 ℃退火30 s,72 ℃ 1 kb/min,35個循環;72 ℃延伸5 min。PCR 結束后取3-5 μL 產物進行瓊脂糖凝膠電泳檢測。將擴增完畢的第二輪PCR 產物混合,取混合后的PCR 產物200 μL,進行瓊脂糖凝膠電泳,切膠回收純化。送北京諾禾致源生物信息科技有限公司進行高通量測序。
1.2.5 脫靶分析
以各基因靶位點序列為種子序列,分別在煙草基因組中數據庫進行Blast比對,在全基因范圍內尋找可能存在的脫靶位點。在候選脫靶位點序列位點上、下游設計引物擴增,通過測序的方法進行驗證。
2 結果與分析
2.1 多基因敲除靶位點的sgRNA設計
利用中國煙草基因組數據庫中PCS1、eIF4E、TOM3、TOM1和HMA2基因的cDNA序列,通過CRISPR MultiTargeter在線軟件對PCS1、eIF4E、TOM3、TOM1和HMA2設計了 CRISPR/Cas9敲除的sgRNA寡聚核苷酸序列,再將設計的各sgRNA序列與基因組DNA進行了聯配分析,篩選出高特異性sgRNA,并排除sgRNA序列夸兩個外顯子的可能性,獲得如表1所示的候選的sgRNA。

表1 多基因敲除的sgRNA寡聚核苷酸序列Tab.1 The sgRNA oligonucleotide sequences of multiple genes
2.2 煙草PCS1、eIF4E、TOM3、TOM1和HMA2基因的sgRNA區域驗證
為了排除基因組測序過程中造成的堿基誤差,確保敲除靶位點序列的特異性和準確性,進一步對含有靶位點sgRNA序列的cDNA片段進行了測序驗證。通過設計特異性擴增引物,以K326品種的cDNA為模板,分別擴增了PCS1、eIF4E、TOM3、TOM1和HMA2含有靶位點序列的cDNA片段,如圖1所示,從K326中均擴增到預期大小的cDNA片段,并進行了純化與克隆。測序分析顯示,PCS1、eIF4E、TOM3、TOM1和HMA2cDNA片段中的sgRNA序列和PAM區與表1中所設計的sgRNA序列及PAM區完全一致,可以作為Cas9對相應基因敲除的sgRNA。
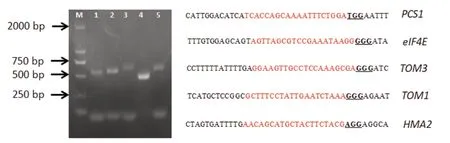
圖1 煙草PCS1、eIF4E、TOM3、TOM1和HMA2 cDNA片段電泳檢測與測序Fig.1 Electrophoresis detection and sequencing of partial cDNA sequence of tobacco PCS1,eIF4E,TOM3,TOM1 and HMA2
2.3 CRISPR/Cas9介導多基因敲除的sgRNA設計與構建
在每個sgRNA上下游分別引入擬南芥U-26啟動子和終止序列,用于獨立啟動和終止sgRNA表達,再將各獨立的sgRNA表達元件依次串聯后,在5'端和3'引入酶切BsaI位點,獲得如圖2A所示的串聯sgRNA片段。利用BsaI酶切的方法將該串聯片段連接至pORE-Cas9植物表達載體上。分別以U26-JC F和PCS1、eIF4E、TOM3、TOM1和HMA2sgRNA的反向引物進行PCR擴增,通過凝膠電泳檢測顯示,5個sgRNA以及載體片段均被擴增到,并呈現出按序排列清晰條帶(圖2B),符合預期目標。這表明已經成功構建了針對PCS1、eIF4E、TOM3、TOM1和HMA2基因敲除的CRISPR/Cas9表達載體。

圖2 多基因敲除的Crispr/Cas9載體構建Fig.2 Construction of Crispr/Cas9 vector for multi-gene knockout
提取重組CRISPR/Cas9載體質粒,采用電擊法轉入農桿菌GV3101,對經過卡那霉素和利福平篩選的單菌落進行菌落PCR驗證,所用引物為U26-JC F和PCS1、eIF4E、TOM3、TOM1和HMA2sgRNA的反向序列。如圖2C所示,得到預期300 bp左右大小的目的條帶,表明重組CRISPR/Cas9載體質粒已經成功轉入農桿菌GV3101菌株內。
2.4 T0代遺傳轉化植株的檢測
利用農桿菌介導的葉盤法將重組CRISPR/Cas9載體質粒轉化煙草K326,經過卡那霉素篩選,獲得了17株T0代遺傳轉化陽性苗。待幼苗長至4~6片葉時,提取葉片基因組DNA,以檢測編碼Cas9蛋白基因的Cas9-JC F和Cas9-JC R引物進行PCR擴增。瓊脂糖凝膠電泳結果顯示,從13株煙草中擴增出預期500 bp左右的Cas9基因片段(圖3),表明構建的重組CRISPR/Cas9基因敲除質粒已成功轉入K326植株中。
2.5 煙草PCS1、eIF4E、TOM3、TOM1和HMA2突變靶位點的擴增與突變分析
設計特異性引物,以各陽性植株DNA為模板,擴增包含突變靶位點在內的150 bp-180 pb的PCR產物。第二輪PCR擴增以第一輪PCR產物為模板,利用Hi-Tom試劑盒進行擴增與加測序接頭的PCR反應,其產物比第一輪長100 bp左右。第一輪和第二輪PCR擴增產物片段大小如圖4所示,純化回收后進行建庫測序。
測序結果分析顯示,PCS1基因在靶位點處發生了多種形式的突變,包括在不同位點處的1個、2個、4個、5個和10個堿基的刪除和1個堿基的插入(圖5A),大多數突變發生在PAM區域附近。Cas9對TOM3基因同樣進行了成功編輯,分析顯示在TOM3-sgRNA序列處主要發生了1個堿基和2個堿基的刪除(圖5 B)。另外,在TOM3-sgRNA位點處分別發現了1個堿基的替換和1個堿基的插入突變。在TOM1-sgRNA序列處有4個不同位點1個堿基的刪除、一個位點2個堿基的刪除以及2個不同位點T→C的堿基替換(圖5C)。在eIF4E-sgRNA位點主要以堿基刪除為主,包括4個不同位點1個堿基的刪除、1個位點4個堿基和1個位點2個堿基的刪除(圖5D),這三種形式的刪除突變均可導致eIF4E基因在煙草中的功能喪失。對HMA2基因的測序結果分析顯示,HMA2-sgRNA位點處不僅有1個堿基的刪除,還存在3個、6個和8個堿基的刪除(圖5E)。根據靶位點中堿基突變的reads與總測序reads的比值,評估了Cas9對靶位點處編輯效率。Cas9對PCS1、TOM3、TOM1、eIF4E、HMA2靶位點處的編輯效率分別約為13.1%、15.1%、6.1%、19.8%和16.8%。
上述分析結果表明,Cas9對以上5個基因均實現了成功編輯,獲得了不同突變類型的T0代植株,堿基突變類型主要以1個堿基的刪除為主,約占總突變類型的50%以上,其次是2個堿基的刪除以及多個堿基的刪除,1個堿基的插入也是導致基因突變的重要因素。在各植株中仍然存在野生型基因序列,因此,T0代突變株為嵌合體。

圖3 T0代植株中Cas9基因片段PCR電泳圖Fig.3 PCR electrophoretogram of the sequence fragments of the Cas9 gene,DL 2000 Marker K326; Wild -type plant of K326;1~13,Transgenic plants

圖4 靶位點序列的PCR電泳圖Fig.4 PCR electrophoretogram of target sequence
對PCS1、eIF4E、TOM3、TOM1和HMA2基因在13棵T0代植株中的突變分布做了維恩分析,如圖6所示,PCS1、eIF4E、TOM3、TOM1和HMA25個基因同時在7棵T0代植株中得到成功突變,這7棵T0代植株分別為 T0-4,T0-5,T0-6,T0-8,T0-9,T0-10和T0-11。5個基因在同一株煙草同時突變的檢出率為53.8%。其余植株中均有不同基因突變的組合,且突變基因數目均在3個或3個以上,而單個基因突變的檢出率在76.9%與92.3%之間,表明多基因敲除系統具有較高的編輯效率。

圖5 Cas9介導煙草PCS1、eIF4E、TOM3、TOM1和HMA2基因突變注:紅色為sgRNA序列,括號中分別標注了突變的堿基與對應的植株編號; WT:野生型Fig.5 Cas9-mediated targeted mutations of PCS1,eIF4E,TOM3,TOM1 and HMA2 in tobacco.Note:sgRNA sequence is marked in red,and the mutated bases and the corresponding plant number are marked in parentheses.WT:Wild Type
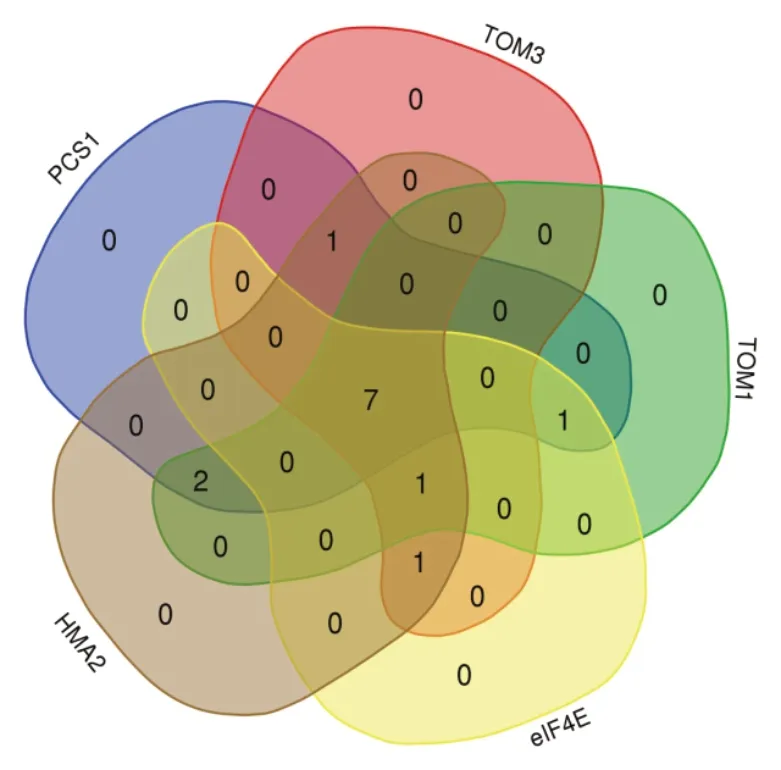
圖6 PCS1、eIF4E、TOM3、TOM1和HMA2基因在T0代植株中的突變分布Fig.6 Mutation distribution of PCS1,eIF4E,TOM3,TOM1 and HMA2 in T0 Generation plants
2.6 脫靶位點預測與檢測分析
由于與靶序列的相似,Cas9可能出現非特異性切割而發生脫靶現象。為了分析本文所構建的CRISPR/Cas9多基因編輯系統是否存在脫靶現象,我們將PCS1、eIF4E、TOM3、TOM1和HMA2基因的靶位點序列與煙草基因組數據進行了對比分析,根據((N)12NGG)的原則和PAM區上游序列(12 bp)決定靶位點特異性的原則[31,32],分別在煙草全基因組范圍內各篩選了一個最有可能脫靶的候選位點,如表2所示,各基因靶位點從第8-10位點開始到PAM序列,完全與脫靶候選位點序列匹配。通過設計特異性引物,在5個基因都有突變的7株陽性植株中擴增候選脫靶位點的DNA片段,測序結果表明并未發現突變序列,這表明本實驗所構建的CRISPR/Cas9系統具有較高的特異性。

表2 CRISPR/Cas9系統脫靶位點分析與檢測Table 2 Analysis and examination of mutations in the putative off-target sites of CRISPR/Cas9 system.
3 討論
CRISPR/Cas9基因編輯系統自出現以來,已在各植物中得到非常廣泛應用。煙草是最早建立CRISPR/Cas9系統的作物之一,Gao等人最早在煙草中利用CRISPR/Cas9系統成功突變NtPDS和NtPDR6基因。隨后,該系統在煙草腋芽、次生代謝、抗逆性等生物學性狀的單基因功能研究方面發揮了重要作用[16-17,33-34]。本文基于煙草中已有CRISPR/Cas9基礎表達系統,首次在煙草中針對不同生物學性狀的多個基因開展了編輯研究。
構建了鎘積累相關基因PCS1和HMA2、PVY抗性相關基因eIF4E、TMV抗性相關基因TOM3和TOM15個靶基因同時敲除的CRISPR/Cas9系統,并轉化至煙草中。通過對陽性植株靶位點的基因序列分析表明,5個靶基因在煙草中均得到了成功編輯,5個基因同時突變的檢出率為53.8%,該比例與水稻中對四個靶位點同時敲除的比例相當(44%和67%)[35],而單基因突變的檢出率在76.9%與92.3%之間,這與煙草單基因突變的陽性植株檢測率相當[16,17]。這表明本文構建的多靶點CRISPR/Cas9系統能夠針對不同性狀的不同基因進行編輯,為煙草不同優良性狀的種質改良提供重要的技術依據。另外,由于普通煙草是由二倍體的絨毛狀煙草和林煙草雜合而來的異源四倍體,在其基因組中有兩套不同二倍體野生煙草基因組類型,因而存在同源基因數量多、基因功能冗余的現象。本文中構建的多靶點的CRISPR/Cas9系統,可有效應用于多個煙草基因的突變,提高煙草功能基因研究效率。
測序結果表明,CRISPR/Cas9系統在煙草中引起的突變,主要是1至10個堿基的刪除,其中1個堿基刪除的突變類型最多,這與前期在煙草和其他植物中所得出的結果一致,這種突變形式很大程度上與植物內在的修復機制相關。另外,由于普通煙草是由二倍體的絨毛狀煙草和林煙草雜合而來的異源四倍體,基因組結構復雜且較大,在T0獲得純合突變的概率不高,多個基因的純合突變篩選需更多的種植代數。CRISPR/Cas9系統引導核酸酶定向剪切活性存在一定程度的容錯概率,會在目標位點序列相似的基因組區域存在非特異剪切活性(脫靶效應)。基因編輯靶位點預測方法較多,全基因組測序是目前被認為最有效的方法。對擬南芥、水稻、棉花中Cas9或Cpf1的脫靶效應研究發現,造成脫靶效應的最主要因素是gRNA特異性,sgRNA嚴謹性設計將有效消除脫靶效應的潛在影響[36-38]。本研究在5個基因同時發生突變的植株中,根據sgRNA特異性識別靶點原則預測和分析了可能的脫靶點,結果表明未發生脫靶現象,與前期在煙草中對CRISPR/Cas9脫靶檢測結果一致[16]。這可能與目標基因的sgRNA均采用嚴謹標準有關,所設計靶序列均是全基因范圍錯誤匹配最低以及GC含量合理的sgRNA。更全面的脫靶評估有待于后續進一步對純合體突變體的全基因組測序分析研究。
4 結論
本研究針對煙草不同性狀的基因構建了多靶點敲除的CRISPR/Cas9系統,獲得了鎘積累相關基因NtPCS1、HMA2、PVY抗性相關基因eIF4E以及TMV抗性相關基因TOM3、TOM1同時突變煙草遺傳材料,5個基因同時突變的檢出率為53.8%。脫靶效應評估分析顯示,在所預測脫靶的候選位點上均未發生脫靶現象。本文構建的多基因編輯的CRISPR/Cas9系統可將煙草多個基因進行有效突變,為煙草基因功能的研究和基于CRISPR/Cas9技術的多性狀改良奠定了基礎。
猜你喜歡
奧秘(創新大賽)(2023年3期)2023-05-06 01:48:20
中國煙草學報(2019年5期)2019-11-14 07:54:12
首都公共衛生(2019年5期)2019-05-21 01:08:34
浙江中西醫結合雜志(2017年2期)2017-01-12 18:23:59
新聞傳播(2016年3期)2016-07-12 12:55:34
當代化工研究(2016年9期)2016-03-20 16:22:08
自動化博覽(2014年6期)2014-02-28 22:32:15
聲屏世界(2014年6期)2014-02-28 15:18:09
西南學林(2013年2期)2013-11-12 12:58:54
中國煙草學報(2012年5期)2012-04-12 06:21:18
