超高效液相色譜-四極桿/靜電場(chǎng)軌道阱高分辨質(zhì)譜測(cè)定蔬菜中的百草枯
2019-09-27 09:22:58邱世婷羅曉梅先正其
分析測(cè)試學(xué)報(bào) 2019年9期
韓 梅,侯 雪,邱世婷,焦 穎,羅曉梅,先正其,王 天
(1.四川省農(nóng)業(yè)科學(xué)院 分析測(cè)試中心,農(nóng)業(yè)部農(nóng)產(chǎn)品質(zhì)量安全風(fēng)險(xiǎn)評(píng)估實(shí)驗(yàn)室(成都),四川 成都 610066;2.賽默飛世爾科技(中國(guó))有限公司,上海 201206)
百草枯又名對(duì)草快,是雜環(huán)類除草劑,屬于吡啶類衍生物,極易溶于水,微溶于醇類及丙酮,不溶于烴類,在酸性及中性溶液中穩(wěn)定,在堿性溶液中易分解[1]。其對(duì)人、畜有較強(qiáng)的急性毒性,且無(wú)特效解毒藥。作為速效滅生性除草劑,百草枯曾廣泛應(yīng)用于農(nóng)業(yè)生產(chǎn),但目前已被20多個(gè)國(guó)家禁用或限用。我國(guó)自2016年7月1日起停止在國(guó)內(nèi)銷售和使用百草枯水劑,2020年9月26日起禁止在境內(nèi)銷售和使用百草枯可溶膠劑[2]。國(guó)家標(biāo)準(zhǔn)GB 2763-2016對(duì)蔬菜、水果、谷物、油料中的百草枯作出了限量要求,規(guī)定各類蔬菜中百草枯允許的最大殘留限量為0.05 mg/kg[3]。由于百草枯的高毒性和廣泛應(yīng)用性,因此建立快速、高選擇性、高靈敏度的百草枯檢測(cè)方法非常必要。
目前有關(guān)百草枯的前處理主要采用固相萃取柱凈化[4],方法較為復(fù)雜;檢測(cè)方法主要有氣相色譜-質(zhì)譜法[5-6]、高效液相色譜法[4,7-9]和高效液相色譜-串聯(lián)質(zhì)譜法[10-13],但上述方法對(duì)復(fù)雜基質(zhì)的抗干擾能力不足,易出現(xiàn)假陽(yáng)性;而靜電場(chǎng)軌道離子阱(Orbitrap)可在很大程度上彌補(bǔ)上述不足。高分辨質(zhì)譜具有較強(qiáng)的抗干擾能力和高通量,且前處理簡(jiǎn)單,能解決基質(zhì)干擾問題。其中,四極桿/靜電場(chǎng)軌道阱高分辨質(zhì)譜(Q-Exactive)是將四極桿與Orbitrap相結(jié)合,不僅具有很好的定性效果,且顯著改善了高分辨質(zhì)譜的定量效果,可與三重四極桿質(zhì)譜相媲美[14-15]。目前將Orbitrap高分辨質(zhì)譜技術(shù)應(yīng)用于蔬菜中百草枯的檢測(cè)尚未見報(bào)道。
本研究建立了蔬菜中百草枯殘留的超高效液相色譜-四極桿/靜電場(chǎng)軌道阱高分辨質(zhì)譜(UHPLC-Q-Exactive MS)分析方法,樣品以乙腈-0.2%甲酸水溶液(1∶1,體積比,下同)提取,分散固相萃取(DSPE)法凈化。該方法前處理簡(jiǎn)單快速、靈敏度高,適用于蔬菜樣品中百草枯的快速檢測(cè)。
1 實(shí)驗(yàn)部分
1.1 儀器與試劑
Q-Exactive 四極桿/靜電場(chǎng)軌道阱高分辨質(zhì)譜儀(配加熱電噴霧離子(HESI)源)、Thermo UltiMate 3000 高效液相色譜儀,配置六通道在線脫氣機(jī)、自動(dòng)進(jìn)樣器(帶大體積進(jìn)樣組件)、柱溫箱(帶2位六通閥)、雙三元梯度泵(美國(guó)Thermo Fisher公司);WH-3微型渦旋混合儀(上海滬西分析儀器廠);TGL-16LM高速離心機(jī)(湖南星科科學(xué)儀器有限公司);AE240電子天平(瑞士Mettler公司)。
標(biāo)準(zhǔn)品:百草枯二氯鹽標(biāo)樣(純度大于99.0%,DR.Ehrenstorfer公司)。
乙腈、甲酸、甲酸銨、乙酸銨(色譜純,美國(guó)Fisher Scientific公司);鹽酸(優(yōu)級(jí)純,四川西隴化工有限公司);十八烷基鍵合硅膠(C18)吸附劑、N-丙基乙二胺(PSA)和石墨化碳黑(GCB)(上海安譜實(shí)驗(yàn)科技股份有限公司);實(shí)驗(yàn)用水為純凈水(由Milli-Q超純水儀制備)。蔬菜樣品來(lái)自四川省農(nóng)貿(mào)市場(chǎng)及蔬菜基地抽檢樣。
1.2 標(biāo)準(zhǔn)溶液的配制
稱取適量百草枯標(biāo)準(zhǔn)品,用0.1 mol/L鹽酸配制成20 mg/L標(biāo)準(zhǔn)儲(chǔ)備溶液,于-4 ℃保存。根據(jù)需要,用試劑或基質(zhì)溶液配制標(biāo)準(zhǔn)儲(chǔ)備溶液,得到不同質(zhì)量濃度的標(biāo)準(zhǔn)工作溶液。
1.3 樣品前處理
準(zhǔn)確稱取5 g(精確至0.01 g)蔬菜樣品于50 mL塑料離心管中,加入10 mL乙腈-0.2%甲酸水溶液(1∶1),渦旋提取1 min后,以4 000 r/min離心5 min,取上層提取液至25 mL容量瓶中,殘留物用10 mL乙腈-0.2%甲酸水溶液(1∶1)重復(fù)提取1次,合并提取液于同一容量瓶中,用水定容至刻度,搖勻。取6 mL提取液于預(yù)先加有300 mg PSA 和300 mg C18的10 mL塑料離心管中,渦旋1 min后,再以4 000 r/min離心力離心5 min。取上清液過(guò)0.22 μm濾膜后,待測(cè)。
1.4 儀器條件
1.4.1 色譜條件色譜柱:Acquity UPLC BEH HILIC柱(100 mm× 2.1 mm,1.7 μm,美國(guó)Waters公司);流動(dòng)相:A為0.2%甲酸水溶液(含50 mmol/L甲酸銨),B為乙腈;流速:0.3 mL/min;進(jìn)樣量:10 μL,柱溫:40 ℃。梯度洗脫程序:0~1.0 min,70%B;1.0~2.0 min,70%~20%B;2.0~4.0 min,20%B;4.0~4.1 min,20%~70%B;4.1~10.0 min,70%B;分析時(shí)間:10.0 min。
1.4.2 質(zhì)譜條件離子源:電噴霧離子源(ESI源);噴霧電壓:3 kV;采集方式:正離子采集模式;檢測(cè)方式:平行反應(yīng)監(jiān)測(cè)(PRM),分辨率:17 500,碰撞能量:21 eV;鞘氣壓力:3.5 MPa,輔助氣壓力:1 MPa;離子傳輸溫度:350 ℃;輔助加熱溫度:400 ℃。
2 結(jié)果與討論
2.1 提取溶劑的選擇
百草枯為堿性離子型化合物,易溶于酸性水溶液,不易溶于乙腈等有機(jī)試劑,但純水溶液以及酸性水溶液對(duì)蔬菜中百草枯的提取效率并不高,而通過(guò)加入一定比例的乙腈可以顯著提高回收率[16]。本研究分別選取水、0.2%甲酸水溶液、2%甲酸水溶液、乙腈、乙腈-0.2%甲酸水溶液(1∶1)以及乙腈-2%甲酸水溶液(1∶1)作為提取溶劑,結(jié)果顯示,乙腈-0.2%甲酸水溶液(1∶1)和乙腈-2%甲酸水溶液(1∶1)為提取溶劑時(shí)百草枯的回收率均超過(guò)90%。考慮到酸度過(guò)大對(duì)儀器、色譜柱性能有影響,因此選擇乙腈-0.2%甲酸水溶液(1∶1)為提取溶劑。進(jìn)一步比較了不同體積比(1∶9、3∶7、1∶1、7∶3、9∶1)的乙腈-0.2%甲酸水溶液的提取效率,發(fā)現(xiàn)兩者比例為1∶1時(shí)百草枯的提取回收率較好且穩(wěn)定,故最終選擇乙腈-0.2%甲酸水溶液(1∶1)為提取溶劑。
2.2 凈化方式的選擇
由于蔬菜基質(zhì)較復(fù)雜,需將提取液進(jìn)一步凈化,從而減少對(duì)儀器的損傷以及基質(zhì)效應(yīng)。本研究采用DSPE技術(shù)進(jìn)行凈化,并對(duì)PSA、C18和GCB凈化劑進(jìn)行考察。其中PSA 能有效去除提取液中的極性物質(zhì)、有機(jī)酸和脂肪酸等雜質(zhì);C18對(duì)親脂性雜質(zhì)和脂肪等具有較好的吸附效果;而GCB可吸附色素、甾醇等大分子非極性干擾物[17]。
比較了無(wú)凈化劑和采用上述不同凈化劑對(duì)百草枯回收率的影響。結(jié)果顯示,不使用凈化劑時(shí)的回收率能達(dá)到分析要求,但雜質(zhì)較多,不利于儀器維護(hù);而單獨(dú)使用1種凈化劑時(shí)的回收率相對(duì)較差。由于本實(shí)驗(yàn)提取出的色素相對(duì)較少,而C18具有一定的去除色素能力,且C18和PSA配合使用對(duì)雜質(zhì)的吸附范圍較廣,因此選用C18和PSA進(jìn)行凈化處理。進(jìn)一步實(shí)驗(yàn)發(fā)現(xiàn),6 mL樣品提取液使用300 mg PSA和300 mg C18即可達(dá)到凈化效果,當(dāng)增加吸附劑用量時(shí),凈化效果并無(wú)明顯改善。因此實(shí)驗(yàn)采用300 mg PSA和300 mg C18進(jìn)行DSPE凈化。
2.3 質(zhì)譜與色譜條件的優(yōu)化
2.3.1 質(zhì)譜條件的優(yōu)化根據(jù)百草枯的結(jié)構(gòu)特征,選擇ESI+作為電離模式。先用全掃描(Full scan)模式找到百草枯的母離子、子離子,然后采用 PRM模式對(duì)百草枯進(jìn)行定性和定量采集。在 PRM 模式下,進(jìn)一步對(duì)二級(jí)質(zhì)譜的碰撞能量(CE)進(jìn)行優(yōu)化,結(jié)果顯示,CE值為21 eV時(shí)能夠獲得最佳的二級(jí)質(zhì)譜定量離子響應(yīng)(圖1A)。由圖1A可知,母離子的精確質(zhì)量數(shù)為m/z186.113 05,二級(jí)質(zhì)譜特征子離子的精確質(zhì)量數(shù)為m/z171.091 19,理論上母離子的分子式為C12H14N2,精確質(zhì)量數(shù)為186.115 15,實(shí)際與理論的精確質(zhì)量數(shù)相差≤5 ppm[18],質(zhì)量準(zhǔn)確度能夠滿足要求。圖1B為百草枯定量子離子的提取離子色譜圖。

2.3.2 流動(dòng)相的選擇參考標(biāo)準(zhǔn)SN/T 0293-2014[4],本實(shí)驗(yàn)選擇乙腈作為流動(dòng)相的有機(jī)相。考察了不同pH值的水系流動(dòng)相(純水、甲酸水溶液)對(duì)百草枯分離效果的影響,結(jié)果表明,不加酸或流動(dòng)相酸度較高時(shí),峰形較差。而在流動(dòng)相中添加0.2%甲酸時(shí),可提高百草枯的離子化效率,分離效果滿意。這是由于流動(dòng)相中加入甲酸,可促進(jìn)化合物的離子化[19];而百草枯為堿性化合物,甲酸能調(diào)節(jié)流動(dòng)相的pH值使之呈酸性,在正離子模式下,流動(dòng)相呈酸性有利于提高百草枯的離子化效率和靈敏度,改善其峰形[20]。因此,選擇乙腈和0.2%甲酸水溶液為流動(dòng)相。
進(jìn)一步比較流動(dòng)相中添加鹽的分離效果,考察了乙腈-0.2%甲酸水溶液、乙腈-0.2%甲酸水溶液(含5 mmol/L乙酸銨)、乙腈-0.2%甲酸水溶液(含5 mmol/L甲酸銨)、乙腈-0.2%甲酸水溶液(含10 mmol/L甲酸銨)、乙腈-0.2%甲酸水溶液(含50 mmol/L甲酸銨)、乙腈-0.2%甲酸水溶液(含100 mmol/L甲酸銨) 6種流動(dòng)相體系。結(jié)果發(fā)現(xiàn),流動(dòng)相中加入銨鹽可改善峰形,且加入甲酸銨比加入乙酸銨的峰形更好,色譜柱達(dá)到平衡狀態(tài)的時(shí)間更短。另外,隨著甲酸銨的濃度升高,峰形變得尖銳,響應(yīng)值增大。當(dāng)甲酸銨濃度為50 mmol/L和100 mmol/L時(shí),峰形和響應(yīng)值無(wú)顯著區(qū)別,但高濃度的甲酸銨會(huì)形成鹽析,損傷色譜柱和管路系統(tǒng)。故最終選擇乙腈-0.2%甲酸水溶液(含50 mmol/L甲酸銨)作為流動(dòng)相。
2.4 基質(zhì)效應(yīng)
本實(shí)驗(yàn)依據(jù)基質(zhì)標(biāo)準(zhǔn)曲線和溶劑標(biāo)準(zhǔn)曲線的斜率比值評(píng)價(jià)基質(zhì)效應(yīng),若斜率比值>1,則表示基質(zhì)對(duì)分析物的響應(yīng)產(chǎn)生增強(qiáng)效應(yīng);若斜率比值<1,則表示產(chǎn)生抑制效應(yīng);若斜率比值為1,則表示不存在基質(zhì)效應(yīng)。當(dāng)斜率比值為0.8~1.2時(shí),表示基質(zhì)干擾程度較低;當(dāng)斜率比值為0.5~0.8或1.2~1.5時(shí),表示有中等程度的基質(zhì)干擾效應(yīng);當(dāng)斜率比值小于0.2或大于1.5時(shí),表示基質(zhì)效應(yīng)干擾強(qiáng)[21]。
采用“1.3”方法得到的空白基質(zhì)提取液和乙腈-0.2%甲酸水溶液(1∶1)分別配制質(zhì)量濃度為0.005~0.5 mg/L的標(biāo)準(zhǔn)溶液,按照本方法進(jìn)行測(cè)定,并繪制基質(zhì)標(biāo)準(zhǔn)曲線和溶劑標(biāo)準(zhǔn)曲線。由表1可見,7種蔬菜中百草枯具有不同程度的基質(zhì)增強(qiáng)效應(yīng)。本實(shí)驗(yàn)采用基質(zhì)匹配標(biāo)準(zhǔn)工作溶液外標(biāo)法進(jìn)行定量,以降低基質(zhì)效應(yīng)的影響。
2.5 線性關(guān)系與檢出限
以“1.3”方法得到的空白基質(zhì)提取液,配制質(zhì)量濃度為0.08~500 μg/L的百草枯基質(zhì)匹配標(biāo)準(zhǔn)溶液。在“1.4”條件下進(jìn)行測(cè)定,以百草枯的質(zhì)量濃度(x,mg/L)為橫坐標(biāo),響應(yīng)峰高(y)為縱坐標(biāo),繪制標(biāo)準(zhǔn)工作曲線。表1結(jié)果表明,百草枯在不同蔬菜基質(zhì)(番茄、辣椒、茄子、黃瓜、菜豆、芹菜、油麥菜)中線性關(guān)系良好,相關(guān)系數(shù)(r2)為0.992 7~0.999 4。并以信噪比S/N≥3 確定儀器的檢出限(LOD),S/N≥10 確定儀器的定量下限(LOQ),得到百草枯的 LOD和LOQ分別為0.1~0.5 μg/kg和0.4~1.8 μg/kg,能夠滿足主要蔬菜品種中百草枯的限量要求[3]。

表1 百草枯在蔬菜中的基質(zhì)效應(yīng)、線性關(guān)系、檢出限和定量下限Table 1 Matrix effects,linear relationships,limits of detection(LOD) and limits of quantitation(LOQ) of paraquat in vegetables
2.6 準(zhǔn)確度與精密度
選用7種代表性蔬菜空白樣品,在優(yōu)化條件下進(jìn)行加標(biāo)回收實(shí)驗(yàn),加標(biāo)水平為50、100、200 μg/kg。由表2可知,百草枯在7種不同蔬菜中的平均回收率為70.8%~113%,相對(duì)標(biāo)準(zhǔn)偏差(RSD)為0.30%~7.9%,方法的準(zhǔn)確度和精密度符合農(nóng)藥殘留分析的要求[18]。圖2為黃瓜基質(zhì)中添加50 μg/kg百草枯的總離子流圖和提取離子流圖。
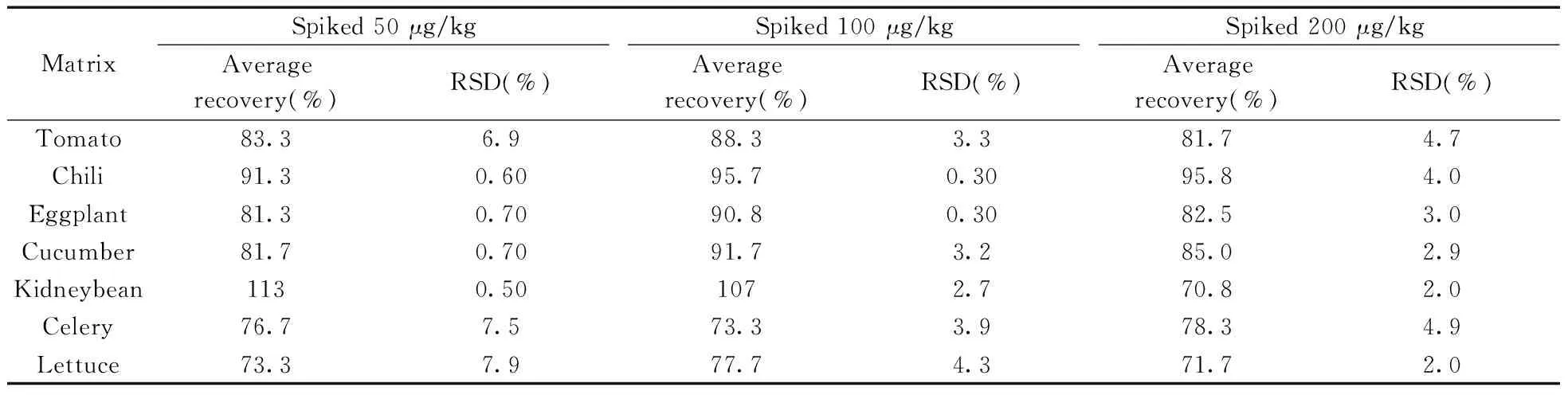
表2 不同加標(biāo)水平下百草枯的平均回收率和相對(duì)標(biāo)準(zhǔn)偏差(n=6)Table 2 Average recoveries and relative standard deviations of paraquat at different spiked levels(n=6)
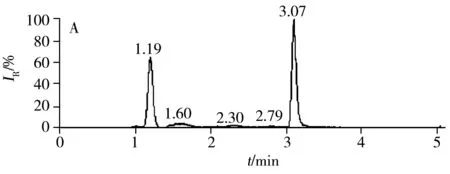
圖2 黃瓜中添加百草枯(50 μg/kg)的總離子流圖(A)和提取離子流圖(B)
Fig.2 Total ion chromatogram(A) and extracted ion chromatogram(B) of paraquat spiked in cucumber(50 μg/kg)
2.7 實(shí)際樣品測(cè)定
應(yīng)用本方法對(duì)四川省多個(gè)地區(qū)隨機(jī)抽取的36個(gè)番茄、33個(gè)茄子、33個(gè)辣椒、41個(gè)黃瓜、10個(gè)菠菜、9個(gè)芹菜、5個(gè)小白菜、5個(gè)萵苣、4個(gè)普通白菜、3個(gè)土豆、2個(gè)蓮花白、2個(gè)蘿卜和2個(gè)芫荽樣品中的百草枯進(jìn)行測(cè)定。結(jié)果顯示,從川西平原抽檢的2個(gè)小白菜、1個(gè)普通白菜、1個(gè)蓮花白和1個(gè)芫荽中檢出百草枯,含量分別為2.06、3.14、3.16、2.31、15.6 μg/kg,說(shuō)明該地區(qū)曾有使用百草枯的現(xiàn)象,具體來(lái)源需進(jìn)一步研究。
3 結(jié) 論
本方法采用乙腈-0.2%甲酸水溶液(1∶1)提取、分散固相萃取凈化,超高效液相色譜-四極桿/靜電場(chǎng)軌道阱高分辨質(zhì)譜快速定性篩查和定量分析蔬菜中的百草枯,利用PRM模式監(jiān)測(cè)其子離子的精確質(zhì)量數(shù),進(jìn)一步降低了背景噪音,從而提高了靈敏度和準(zhǔn)確性。結(jié)果表明,蔬菜中百草枯的檢出限為0.1~0.5 μg/kg,定量下限為0.4~1.8 μg/kg,平均回收率為70.8%~113%,相對(duì)標(biāo)準(zhǔn)偏差為0.30%~7.9%。該方法前處理操作簡(jiǎn)單快速,靈敏度高,準(zhǔn)確性好,可用于主要蔬菜品種中百草枯的檢測(cè)。

- 分析測(cè)試學(xué)報(bào)的其它文章
- 基于微波消解/電感耦合等離子體質(zhì)譜法測(cè)定土壤中全磷
- QuEChERS/高效液相色譜-串聯(lián)質(zhì)譜法測(cè)定面膜中10種熒光增白劑
- 通過(guò)式固相萃取/超高效液相色譜-串聯(lián)質(zhì)譜法快速測(cè)定動(dòng)物源性食品中安乃近代謝物殘留量
- 超高效液相色譜-四極桿/靜電場(chǎng)軌道阱高分辨質(zhì)譜快速分析化妝品中17種喹諾酮類藥物
- 水產(chǎn)品中11種海洋生物毒素的高效液相色譜-四極桿/靜電場(chǎng)軌道阱高分辨質(zhì)譜檢測(cè)方法研究
- 桃中農(nóng)藥殘留分析及膳食暴露評(píng)估研究