超高效液相色譜-串聯質譜快速測定飲料中2種生物堿和6種甜味劑
2019-10-09 03:07:36劉紅王林胡博李麗霞黃偉劉蕓
食品與發酵工業 2019年17期
劉紅,王林,胡博,李麗霞,黃偉,劉蕓,3*
1(湖北省藥品監督檢驗研究院,湖北 武漢,430075)2(湖北中醫藥大學,湖北 武漢,430065)3(武漢大學 化學與分子科學學院,湖北 武漢,430072)
目前市場的飲料品種繁多,為了使其具有更好的口感和更好的功能作用,不法商家違規濫用、超范圍使用添加劑。咖啡因與茶堿屬于生物堿類,可樂型碳酸飲料、茶、咖啡及植物類飲料中含有上述一種或兩種成分。適量的攝入,可緩解緊張情緒,改善血管功能,利尿,擴血管;過量攝入,則會產生心動過速、高血壓、胃病、失眠等不良反應[1]。糖精鈉、安賽蜜、阿斯巴甜、紐甜和三氯蔗糖是飲料中常用的非糖類甜味劑,一般甜度很高,用量極少[2]。2017年10月31日《關于愛德萬甜等6種食品添加劑新品種、環己基氨基磺酸鈉(又名甜蜜素)等6種食品添加劑擴大用量和使用范圍》的公告(2017年第8號)中增補新型甜味劑愛德萬甜[3],在茶、咖啡、植物(類)飲料中的限量為0.003 g/kg,固體飲料的限量為0.004 g/kg。
咖啡因和非糖類甜味劑檢測的國家標準[4-9]采用不同的檢測方法。糖精鈉、安賽蜜、阿斯巴甜、紐甜、咖啡因和三氯蔗糖均采用高效液相色譜法,其中,糖精鈉、安賽蜜、阿斯巴甜、紐甜和咖啡因使用紫外檢測器或二極管陣列檢測器,三氯蔗糖使用蒸發光散射檢測器,且樣品的預處理方法各不相同。愛德萬甜和茶堿目前暫無國標方法,愛德萬甜主要采用高效色譜法和高效液相串聯質譜法[10-11],茶堿主要采用高效色譜法[12]。可見,目前檢測上述幾種成分主要采用高效液相色譜法,且均只針對某種或某幾種成分的檢測,尚未見對同一樣品中2種生物堿和6種甜味劑同時進行測定的報道。采用高效液相色譜法可能會遇到以下問題:(1)僅靠保留時間對色譜峰定性,對保留時間相近的色譜峰定性不準確;(2)很難排除復雜基質樣品中基質對成分的干擾。因此,建立一種能同時測定這些成分,且定性定量準確的方法十分必要。本實驗超高效液相色譜-串聯三重四極桿質譜(ultra-performance liquid chromatography-mass spectrometry/mass spectrometry,UPLC-MS/MS),能在較短的時間內對2種生物堿和6種甜味劑進行一次性的分離和測定,填補了飲料中新型甜味劑愛德萬甜和2種生物堿同時測定的空白。
1 材料與方法
1.1 材料與試劑
甲醇、乙腈(色譜純),默克股份兩合公司;甲酸銨、甲酸(色譜純),Aladdin公司;乙酸銨(色譜純),Macklin公司。愛德萬甜(含量≥97.0%),美國SIGMA公司;糖精鈉(1.0 g/L),中國計量科學研究院;安賽蜜(1.0 g/L),農業部環境保護科研監測所;三氯蔗糖(含量為98.5%),德國Dr Ehrenstorfer公司;阿斯巴甜(含量為97.7%),德國Dr Ehrenstorfer公司;紐甜(含量100%),美國stanford公司;咖啡因(含量為100%)、茶堿(含量為100%),中國檢驗測疫科學研究院。
10批樣品(3批茶飲料和7批果蔬汁),均來源2018年湖北省抽檢樣品。
1.2 儀器與設備
XEVOTMTQ-S液相色譜-串聯質譜,美國Waters公司;Milli-Q超純水器,美國Millipore公司;LC-250超聲波清洗機,山東濟寧魯超超聲設備有限公司;臺式離心機,德國Thermo Biofuge公司;ACQUITY UPLC?BEH C18,美國Waters公司;XP504電子天平,瑞士梅特勒公司。
1.3 方法
1.3.1 標準溶液的配制
分別精密稱取一定量各標準物質于棕色容量瓶中,加甲醇溶解并定容至刻度,配制成質量濃度為10 mg/L標準儲備溶液。臨用時,用空白樣品提取液逐級稀釋,根據每種目標化合物的定量限,配制成不同濃度的系列基質混合標準工作液。
1.3.2 前處理方法
精確稱取1.0 g樣品置于10 mL比色管中,加入V(乙腈)∶V(水)=20∶80混勻,定容至刻度,超聲10 min,以10 000 r/min離心5 min,上清液過0.22 μm有機濾膜,待測。
1.3.3 色譜-質譜條件
色譜柱:ACQUITY UPLC?BEH C18(1.7 μm,2.1 mm×50 mm);進樣量2 μL;柱溫35 ℃;流速0.3 m/min;流動相:A相為0.1%甲酸溶液,B相為乙腈;流動相梯度洗脫程序:0~2.00 min,97%A~90%A;2.00~8.00 min,90%A~60%A;8.00~10.00 min,60%A~10%A;10.00~10.10 min,10%A~97%A;10.10~13.00 min,97% A;離子源:電噴霧離子源(ESI);掃描方式:正負離子掃描;掃描模式:多反應監測(MRM);毛細管電壓3.5 kV;去溶劑溫度350 ℃;去溶劑氣流速800 L/Hr。詳細的質譜參數列于表1。
2 結果與分析
2.1 樣品提取的優化
8種目標化合物中,咖啡因和愛德萬甜更易溶于有機溶劑,其他6種易溶于水,所以選擇了乙腈∶水的體積比分別為:2∶8、5∶5、8∶2等3種提取液,對8種目標化合物的提取回收率進行比較。結果表明,阿斯巴甜、紐甜、愛德萬甜、三氯蔗糖和咖啡因3種溶劑提取回收率相近。當提取溶劑中乙腈體積分數為80%時,糖精鈉,安賽蜜的回收率顯著下降;當提取溶劑中乙腈體積分數≥50%時,茶堿回收率顯著下降。回收率下降的原因可能是3種物質均為水溶性,有機相比例增大時,降低了提取效率。故本文選用V(乙腈)∶V(水)=2∶8作為提取溶劑。
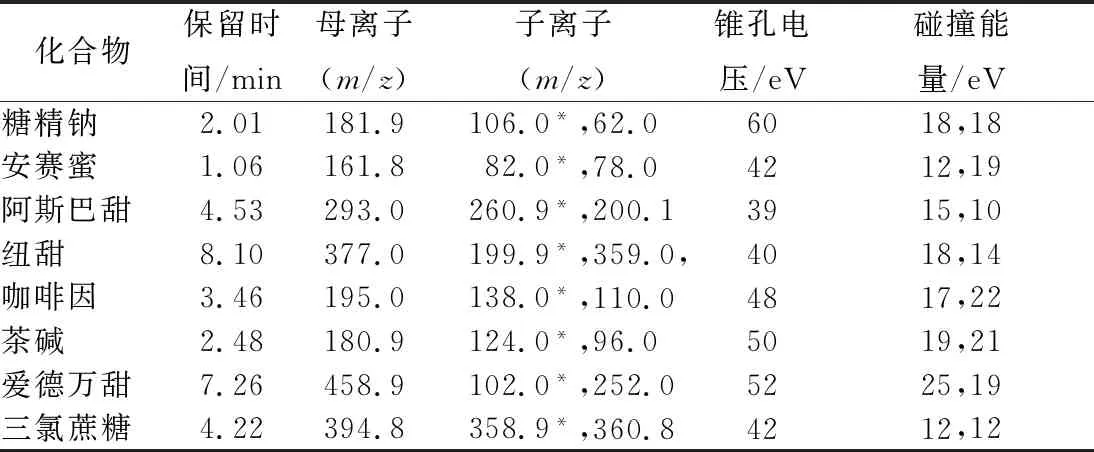
表1 八種化合物的保留時間和質譜參數Table 1 Retention times and MS parameters for 8 target compounds
注:*表示定量離子對。
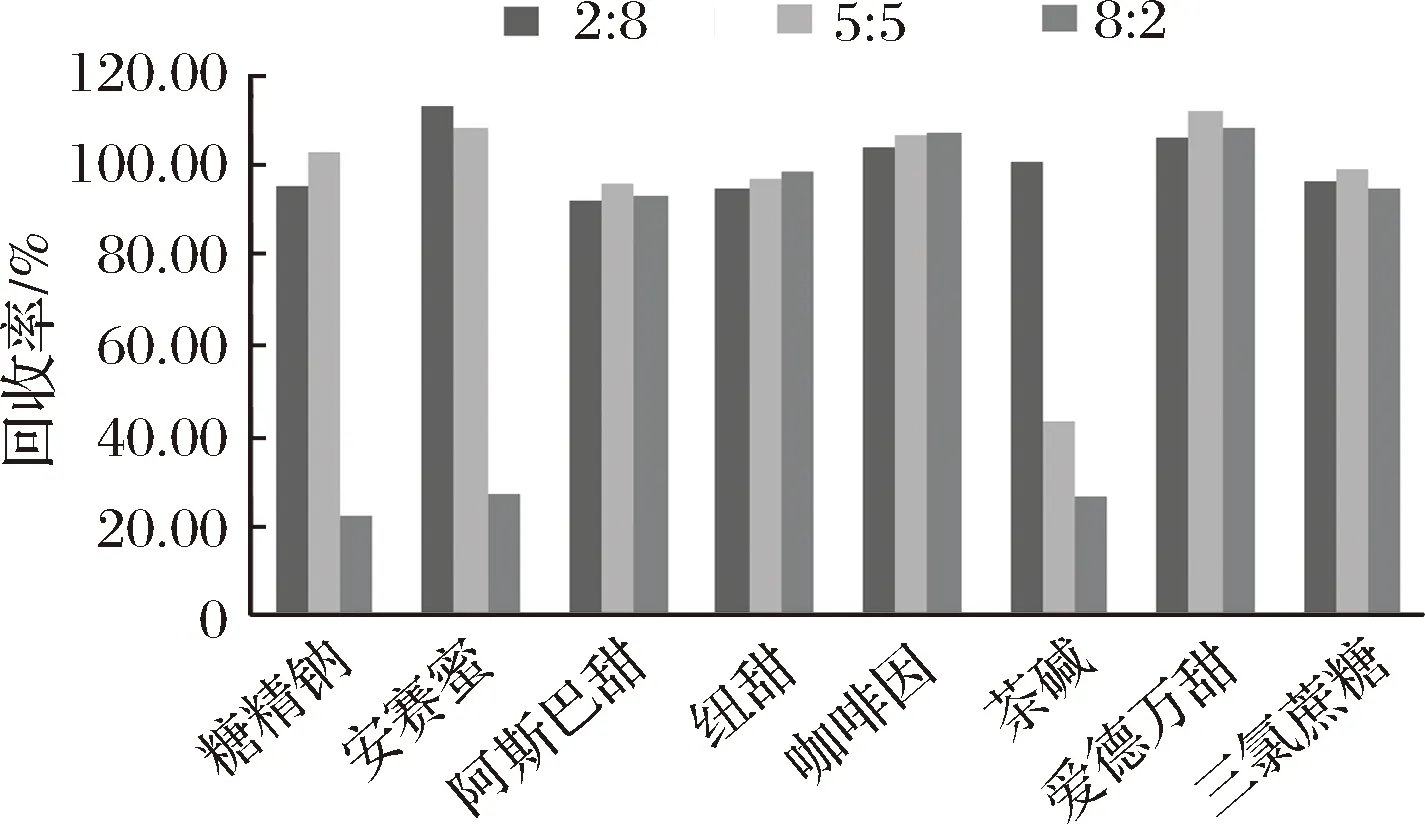
圖1 三種提取溶劑提取回收率的比較Fig.1 Comparison of the recovery of three extracting solvents
2.2 色譜條件的優化
流動相的選擇對于目標物的分離、靈敏度以及峰形有較大的影響。本文考察了乙腈-0.1%甲酸溶液、乙腈-0.01 mol/L甲酸銨溶液、乙腈-0.01 mol/L乙酸銨溶液(含0.05%甲酸)和乙腈-0.01 mol/L乙酸銨溶液四種流動相體系,見圖2。
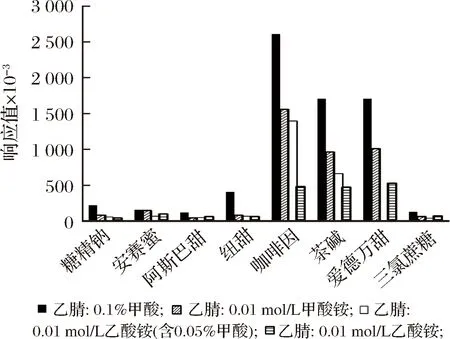
圖2 四種流動相體系的比較Fig.2 Comparison of four mobile phase systems
結果表明,以乙腈-0.1%甲酸溶液作為流動相體系時,8種目標化合物的響應均優于其他流動相體系,且峰形較好。故選擇乙腈-0.1 %甲酸溶液作為流動相體系。
2.3 質譜條件的優化
分別在正離子和負離子模式下對各質量濃度為200 ng/mL的目標化合物單一對照溶液進行全掃描,選出合適的電離方式及母離子;在確定ESI離子監測模式后,分別以分子離子為母離子,對其子離子進行全掃描,選取豐度較強及干擾較小的2個子離子分別作為定量離子和定性離子;以子離子掃描(daughter scan)方式,優化了目標化合物二級質譜的碰撞能量等質譜分析參數(表1),色譜圖見圖3。
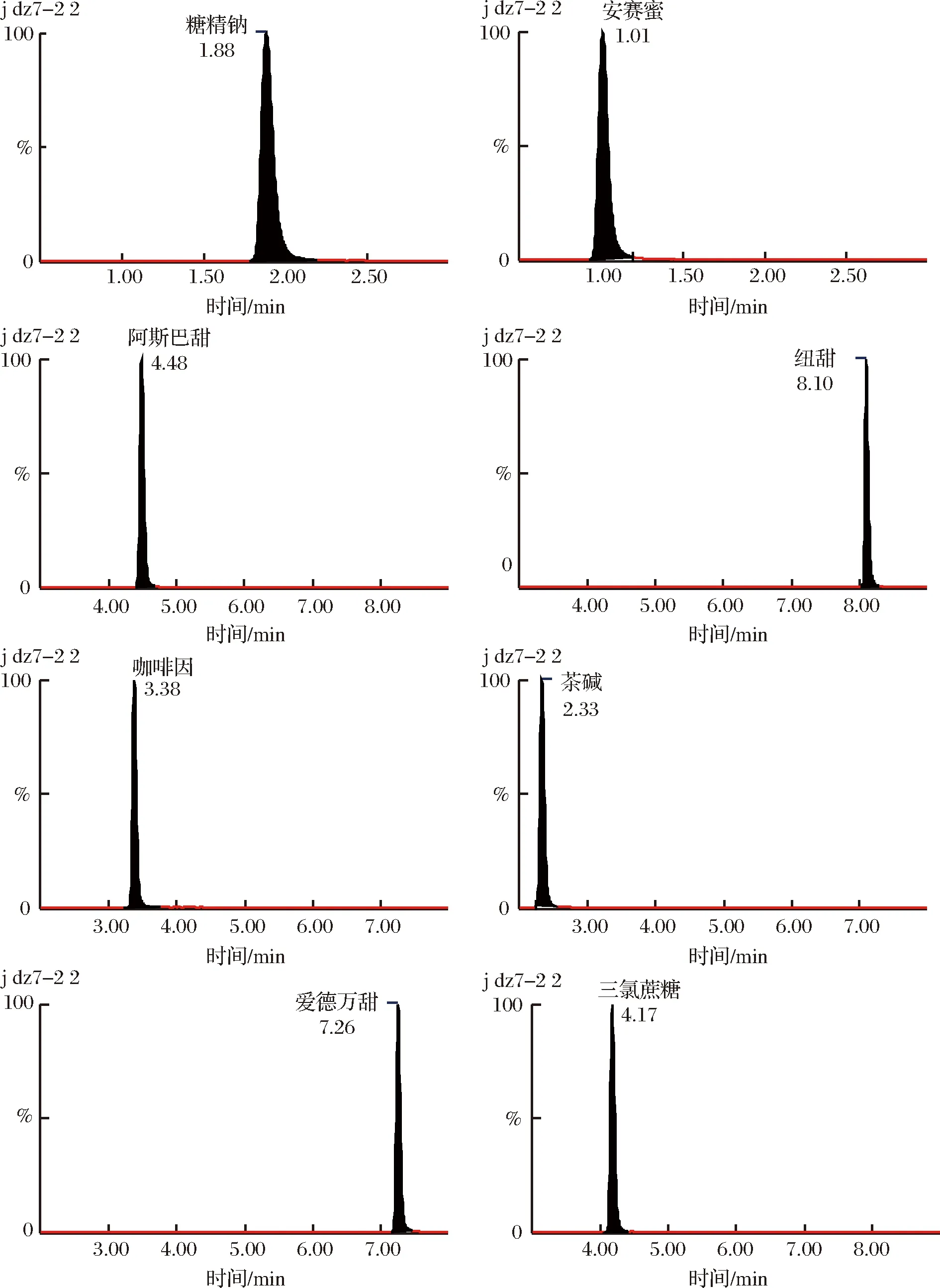
圖3 混合標準樣品的定量離子色譜圖Fig.3 Quantitative ion chromatograms of mixed standards
2.4 基質效應(matrix effect,ME)
評價基質效應是指樣品中除被分析物以外的化合物在分析過程中對目標物有顯著干擾,并影響結果準確性的現象。以典型樣品果蔬汁為樣品基質,建立的快速測定方法中雖然采用V(乙腈)∶V(水)=20∶80提取溶液可以沉淀部分蛋白質,但未對樣品溶液進行凈化處理,可能存在一定的基質效應,所以采用基質標準曲線與溶劑標準曲線的斜率百分比和樣品加標回收率兩種方式來評價基質效應(ME)[13-15]。當ME>100%時,基質存在離子化增強作用;當ME<100%時,基質存在離子化抑制作用。結果見圖4。
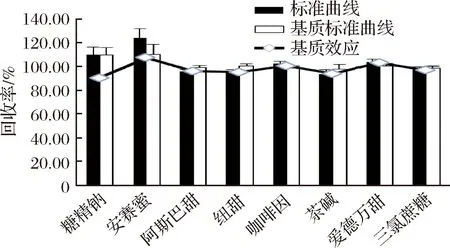
圖4 飲料基質對8種目標化合物的基質效應Fig.4 Matrix effect of 8 target compounds in drink
由圖4可見,安賽蜜、咖啡因和愛德萬甜存在離子化增強作用,糖精鈉、阿斯巴甜、紐甜、茶堿和三氯蔗糖存在離子化抑制作用。由基質標準曲線和溶液標準曲線分別測定的加標回收率可見,咖啡因和三氯蔗糖的溶液標準曲線回收率略優于基質標曲回收率,其他6種目標測定物基質回收率均優于溶液標準曲線回收率。雖然基質效應值在85%~115%,為了獲取更準確的定性、定量結果,綜合考慮加標回收率結果,本文采用系列基質標準工作液進行分析。
2.5 檢出限、定量限和線性范圍
用飲料空白樣品按照1.3節前處理方法得到樣品提取液,根據目標化合物的質譜響應強弱配制成不同濃度的混合標準溶液系列、以目標化合物的色譜峰面積Y對其質量濃度x(μg/mL)進行線性回歸,用空白基質提取液稀釋標準溶液,直到獲得每種目標化合物的信噪比S/N≥3和S/N≥10的濃度,確定其為該化合物的檢出限和定量限,結果見表2。
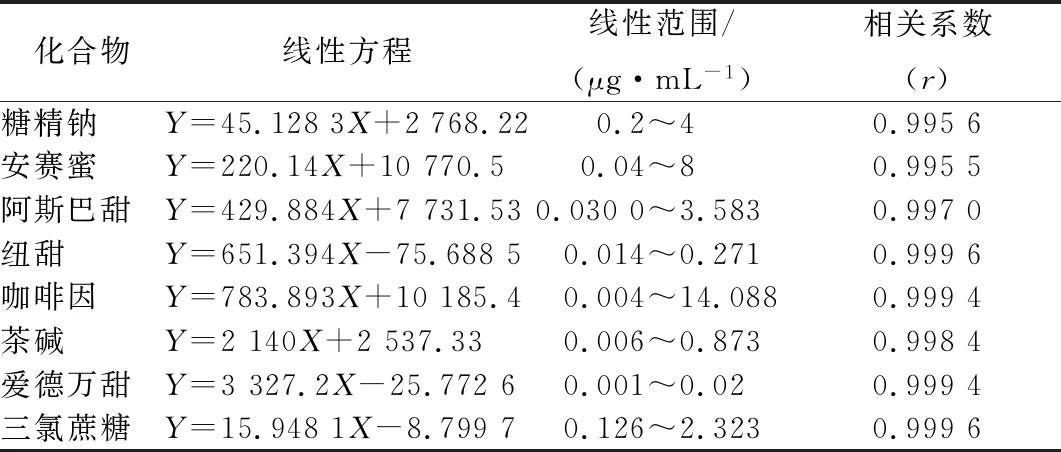
表2 線性方程、線性范圍及相關系數Table 2 Linear equations,linear ranges and correlation coefficient
飲料中各目標化合物在相應的濃度范圍內,線性關系良好,相關系數均大于0.995,檢出限為0.009~0.5 mg/kg,定量限均為0.03~2 mg/kg。與咖啡因和非糖類甜味劑檢測的國家標準相比較,其靈敏度顯著提高(表3)。
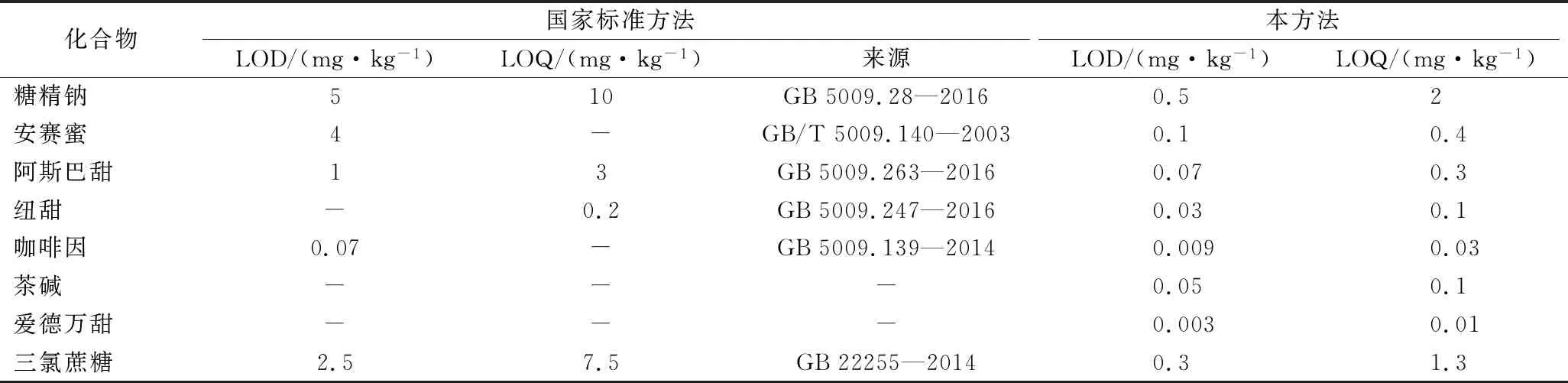
表3 本方法與國標方法的LODs和LOQs比較Table 3 Comparison of LODs and LOQs with the method and national standards
注:“-”表示沒有檢出。
2.6 回收率與精密度
在空白飲料樣品添加8種標準溶液,添加水平為LOQ、3LOQ和5LOQ,每個添加水平平行分析6次,平均回收率為92.3%~110.4%,相對標準偏差(RSD)為0.9%~10.8% (n=6)(見表4)。

表4加標樣品的回收率和相對標準偏差(n= 6)Table 4 Recoveries and RSDs of target compoudsad- vantame in spiked samples
2.7 實際樣品測定
采用本文建立的方法對市售的10個飲料樣品進行前處理(3個茶飲料,7個果蔬汁飲料),并采用超高效液相色譜-串聯質譜進行了檢測,其中3個樣品檢出咖啡因和茶堿,1個樣品檢出安賽蜜和阿斯巴甜,其他成分未檢出,結果見表5。
3 結論
本文建立了超高效液相色譜-串聯三重四極桿質譜快速測定飲料中2種生物堿和6種甜味劑的方法。以V(乙腈)∶V(水)=20∶80超聲提取樣品后,用超高效液相色譜-串聯質譜分析檢測,并進行了方法學驗證和10個實際樣品的測定。該方法簡單、快速,靈敏,準確,其定量限低于國家標準,適用于飲料中2種生物堿和6種甜味劑檢測,填補了飲料中新型甜味劑愛德萬甜和2種生物堿同時測定的空白,可為食品安全監管提供有效保障。

表5 樣品測定結果表 單位:mg/kgTable 5 The results of the samples
注:ND表示沒有檢出。
