改進QuEChERS-氣相色譜-質譜法測定蘋果中胺鮮酯殘留
2019-11-07 01:03:26孔祥吉張雪梅孔德洋
色譜 2019年12期
關鍵詞:方法
孔祥吉, 孔 順, 張雪梅, 孔德洋
(1.生態環境部部南京環境科學研究所, 江蘇 南京 210042; 2.東南大學土木工程工程學院, 江蘇 南京 211189; 3.江蘇省計量科學研究院, 江蘇 南京 210023)
胺鮮酯(diethyl aminoethyl hexanoate, DA-6),化學名稱為乙酸二乙氨基乙醇酯,是一種新型、廣譜性植物生長調節劑[1]。研究表明,低濃度胺鮮酯對多種植物有調節、控制和促生長的作用[2],被廣泛應用于玉米、白菜、花生、水稻等多種農產品中[3,4]。同時,為防止其過量使用導致的農殘問題,必須制定相應的殘留限量標準。我國現行的《食品中農藥最大殘留限量》(GB 2763-2016)僅規定了胺鮮酯在玉米、花生仁、普通白菜和小白菜中的臨時限量,且沒有相應的標準方法。
制定食品中農藥的殘留限量,重要的依據就是檢測方法的檢出限。目前,國內報道的胺鮮酯的測定方法主要是氣相色譜法,內容偏重于原藥產品純度的分析[5,6],此外,采用氣相色譜-質譜(GC-MS)、液相色譜-質譜(LC-MS)對水、土壤、水溶性肥料和白菜等環境樣品中胺鮮酯殘留檢測的方法偶有報道[7-10]。但未見復雜基質如高糖或高色素量的水果中胺鮮酯殘留分析的研究。
國內已有使用胺鮮酯提高蘋果品質的實例[11],制定蘋果中胺鮮酯殘留限量的要求日益緊迫。近年來,果蔬中農藥殘留的分析主要是通過QuEChERS結合色譜-質譜技術,但蘋果中胺鮮酯的測定存在一定難度:一是基質復雜,二是該農藥的理化性質尚未有明確報道。本文采用GC-MS法,并對QuEChERS前處理過程進行優化,同時以QuEChERS結合超高效液相色譜-串聯質譜(UPLC-MS/MS)對樣品添加過程進行驗證。本實驗建立的分析方法針對性強,靈敏度高,適用于蘋果中胺鮮酯殘留的測定,可為該農藥殘留限量標準的制定提供參考。
1 實驗部分
1.1 儀器、試劑與材料
7890A-5975C氣相色譜-質譜儀(美國Agilent公司); Agilent Technologies 1290 Infinity超高效液相色譜-AB SCIEX Triple Quad 4500三重四極桿質譜儀(美國AB公司); A11基本型分析研磨機、T18均質儀(德國IKA公司); Centrifuge 5804高速離心機、10 μL~1 mL移液槍(德國Eppendorf公司); R-210旋轉蒸發儀(瑞士Buchi公司); Milli-Q超純水機(美國Millipore公司);電子天平(0.1 mg,瑞士Mettler Toledo公司)。
胺鮮酯標準品(純度≥98.5%,國家標準物質研究中心);乙腈、二氯甲烷、丙酮(色譜純,德國Merck公司);氯化鈉、無水硫酸鎂、無水硫酸鈉(分析純,南京化學試劑有限公司);乙二胺-N-丙基硅烷(PSA,分析純,美國Waters公司)實驗用水為Milli-Q(美國Millipore公司)制備的超純水。
供試的蘋果樣品為市場上購買的有機蘋果,經研磨成漿后冷凍保存。
1.2 標準溶液配制
稱取10.2 mg胺鮮酯標準品,置于100 mL棕色容量瓶中,用丙酮溶解并定容至刻度,配制成0.1 g/L的標準儲備液,于4 ℃避光保存,有效期3個月。分別移取適量標準儲備液,用丙酮稀釋,配制0.005~0.2 mg/L系列標準溶液,供GC-MS檢測。若采用UPLC-MS/MS儀器檢測,則使用乙腈作溶劑,標準品稱量和溶劑用量與采用GC-MS時相同。
1.3 GC-MS分析條件
色譜柱:Agilent HP-5 MS柱(30 m×250 μm×0.25 μm);進樣口溫度:260 ℃;載氣:氦氣(純度≥99.999%);流速:1.5 mL/min。升溫程序:初始溫度80 ℃,保持0.5 min;以15 ℃/min升溫至120 ℃,保持1 min;以5 ℃/min升溫至160 ℃,保持2 min;再以25 ℃/min升溫至200 ℃,保持1 min;最后以50 ℃/min升溫至280 ℃,保持2 min。進樣方式:不分流進樣;進樣量:1 μL。
離子源:EI源;離子源溫度:230 ℃;四極桿溫度:150 ℃;電子掃描方式:選擇性離子掃描(SIM)。特征碎片:m/z86、100和143;定量碎片:m/z86。
1.4 UPLC-MS/MS分析條件
色譜柱:Agilent EC-C18柱(75 mm×2.1 mm, 2.7 μm);柱溫:30 ℃;流動相A:甲醇;流動相B: 0.2%(v/v)甲酸水溶液;流速:0.4 mL/min。梯度洗脫程序:0~0.01 min, 50%B; 0.01~1.00 min, 50%B~25%B; 1.00~2.00 min, 25%B~50%B; 2.00~4.50 min, 50%B~90%B; 4.50~5.00 min, 90%B~50%B。進樣量:5 μL。
離子源:ESI源,正離子模式;掃描方式:多反應離子監測(MRM);定性/定量離子對:m/z216.1/143.3和216.1/99.9;去簇電壓(DP): 40 V;碰撞能量(CE): 23.6 eV(定性離子對)、22.0 eV(定量離子對);入口電壓(EP): 10 V;碰撞室出口電壓(CXP): 7.0 V。
1.5 樣品前處理
1.5.1對應GC-MS的操作
準確稱取磨碎后的供試蘋果樣品5 g(精確至0.01 g),置于80 mL具塞離心管中,加入25 mL加熱至40 ℃的超純水,渦旋30 s,勻漿2 min。勻漿后的混合物經高速離心分離后過濾,將濾液轉移至分液漏斗中,用20 mL二氯甲烷液液萃取,重復萃取1次,取混合后的萃取液10 mL,于雞心瓶中緩慢旋蒸至干,加入2 mL丙酮溶解殘余物并定容。定容液轉移至10 mL離心管中,加入0.1 g PSA和0.1 g無水硫酸鎂,渦旋30 s,以8 000 r/min離心5 min,取1 mL離心后的溶液,過0.22 μm有機濾膜,供GC-MS檢測。
1.5.2UPLC-MS/MS對加標回收率的驗證
同時,建立胺鮮酯分析的超高效液相色譜-串聯質譜法,相應地,將1.5.1樣品前處理最后一步的定容溶劑換作乙腈,對方法的回收率進一步確定。
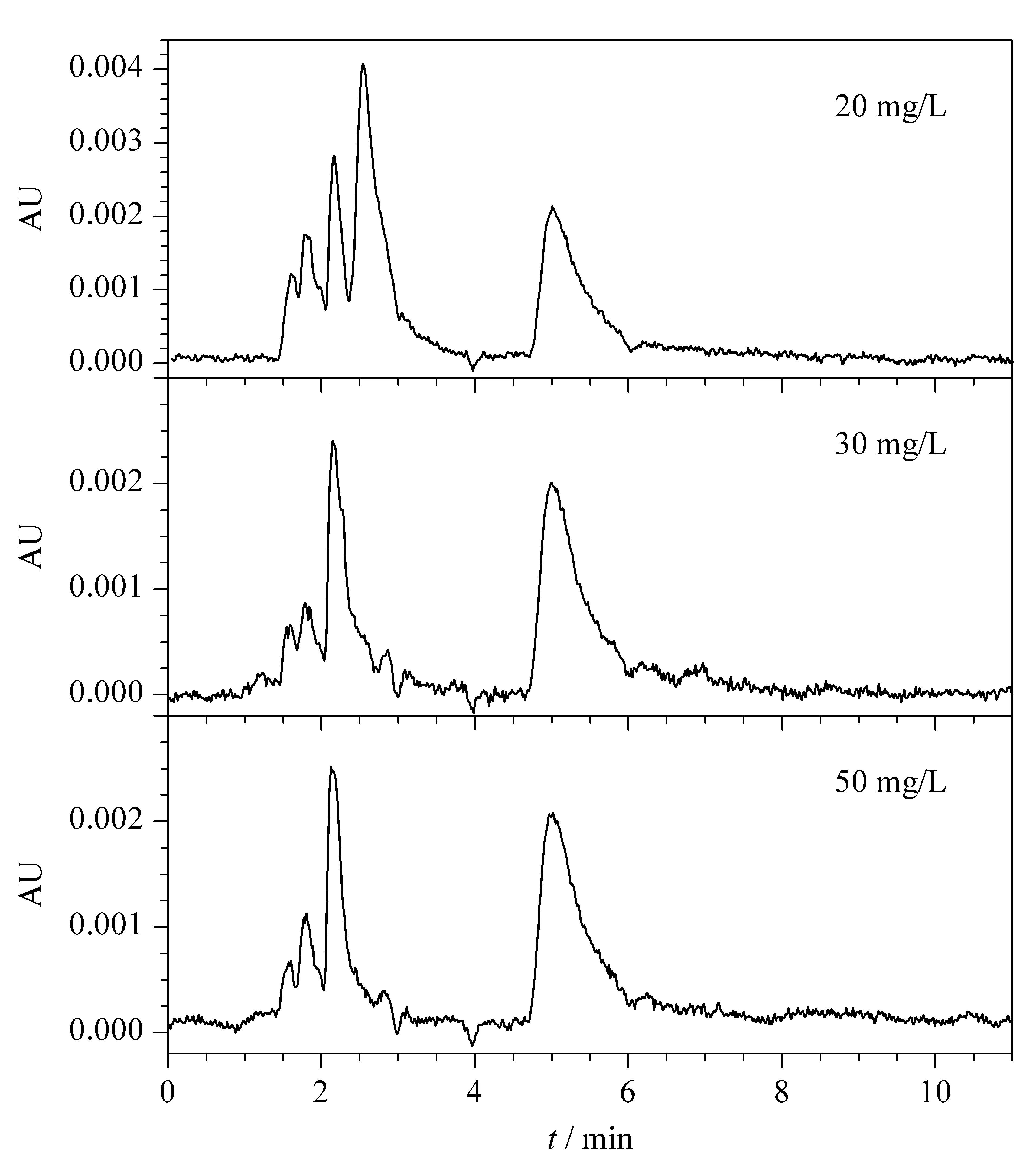
圖1 不同含量的胺鮮脂標準溶液的色譜圖(210 nm)
2 結果與討論
2.1 儀器方法的建立
2.1.1高效液相色譜-光電二極管陣列檢測的可行性
為探究使用HPLC測定胺鮮酯的可行性,本實驗配制20、30和50 mg/L胺鮮脂的乙腈標準溶液,以甲醇-0.1%(v/v)磷酸水(7∶3, v/v)作為流動相,采用RP-C18色譜柱(250 mm×4.6 mm, 5 μm),在柱溫30 ℃下,用光電二極管陣列檢測器(全掃描波長)分析。結果表明:在190~400 nm波長下,獲得的色譜圖中均無胺鮮脂的特征吸收峰(見圖1)。可能是胺鮮酯的分子結構(見圖2)中不具備對光電二極管陣列檢測器響應的基團。胺鮮脂的分子結構中僅有一個羧基,不具備通常產生紫外吸收的C=C共軛及苯環結構,而羧基的吸收波長多為100~200 nm。因此高效液相色譜-光電二極管陣列檢測器無法檢測胺鮮脂。

圖2 胺鮮酯的分子結構圖
2.1.2GC-MS方法的優化
參考已有文獻[7],初步建立了GC測定胺鮮酯的方法。同時,為避免蘋果中雜質分子的干擾,對升溫程序進一步優化,使其保留時間(9.2 min)在合理范圍內,保證胺鮮酯既不受雜質峰的干擾,也不因保留時間滯后降低分析效率。
色譜柱的選擇 通常有機農藥的分析采用HP-1 MS或HP-5 MS色譜柱,HP-1 MS色譜柱的填充物為二甲基硅氧烷,通常針對非極性化合物有較好的分離效果,應用于相對分子質量較低的醇類,分析效果較佳;HP-5 MS柱的填充物為5%二苯基-95%二甲基亞芳基硅氧烷共聚物,熱穩定性較高,柱流失低,適合弱極性化合物的分離。因此,本研究選擇HP-5 MS色譜柱。
特征碎片離子的選擇 首先采用全掃描模式,對0.2 mg/L胺鮮酯的丙酮標準溶液進行測定,確定其特征碎片離子的m/z為86、100和143;同時發現m/z為86的碎片離子豐度值較大且穩定,宜作為定量離子。
進樣溶劑的影響 胺鮮酯在乙酸乙酯、二氯甲烷及丙酮溶液中均具有較好的溶解性。通過多次進樣,發現乙酸乙酯作進樣溶劑時,其在色譜柱、進樣針或進樣口墊片處可能存在較大殘留,導致重復性測定結果不穩定,同一樣品多次進樣的峰面積值浮動高達200%。從溶劑極性和對HP-5 MS色譜柱填充物保護的角度考慮,相比二氯甲烷,丙酮作為進樣溶劑更為合適。在優化的分析條件下,胺鮮酯標準溶液(0.1 mg/L)的色譜圖見圖3。

圖3 胺鮮脂標準溶液(0.1 mg/L)的色譜圖
2.2 前處理方法的優化
QuEChERS是一種農藥多殘留前處理方法。該方法操作簡單,前處理時間短,有機溶劑用量少,已成為多殘留分析的首選方法[12,13]。為探究契合GC-MS方法的樣品前處理方法,本實驗對比了0.008、0.01和0.1 mg/kg添加水平下,3種前處理方法下蘋果中胺鮮脂的加標回收率。
方法一:將提取劑丙酮、凈化劑無水硫酸鎂和輔助提取劑氯化鈉,加入到研磨后的蘋果中進行勻漿提取,提取后的混合物經高速離心分離、過濾,濾液再以PSA+無水硫酸鎂凈化,凈化液在1.3節條件下測定。
方法二:將提取劑乙腈(或丙酮)、凈化劑無水硫酸鎂和輔助提取劑氯化鈉加入到研磨后的蘋果中勻漿提取,提取后的混合物經高速離心分離、過濾,濾液經旋轉蒸發濃縮并以丙酮定容,定容液以PSA+無水硫酸鎂凈化,凈化液在1.3條件下測定。
方法三:同1.5.1節描述。
采用方法一:在0.008、0.01和0.1 mg/kg 3個添加水平下,樣品中胺鮮酯的峰面積平均值均在106以上,且與添加水平無明顯正相關性。0.01 mg/kg添加水平(理論進樣濃度為0.002 5 mg/L)下的峰面積甚至高于0.02 mg/L的基質溶液。分析其中原因,可能主要是因為丙酮提取液中雜質成分較多,雜質峰對樣品峰產生包裹作用;抑或是在溶劑作用下,胺鮮酯分子與雜質分子形成了配合物,且不能通過凈化作用去除。有研究[13]將農藥的乙腈提取液經凈化后直接進GC-MS系統檢測,但本研究認為,乙腈膨脹系數大,其溶液在汽化時容易充滿襯管造成目標物不能全部進入色譜柱,引起檢測誤差或污染系統,因此未采用。
采用方法二:在0.1 mg/kg添加水平下,無論以丙酮作提取溶劑,還是以乙腈作提取溶劑,胺鮮酯的平均回收率均低于10%。造成該結果可能的原因是在旋轉蒸發濃縮步驟中,提取液基質作用產生的影響,由于受提取溶液中果糖、蛋白質等大分子影響,提取液沸程增長,導致具有一定揮發性的胺鮮酯產生較大的損失,回收率過低。
采用方法三:基質作用對測試結果的影響在可接受范圍內,在0.01 mg/kg和0.1 mg/kg添加水平下,胺鮮酯的平均回收率≥75.0%, RSD<3.0%,滿足測定要求。分析其原因,可能得益于反萃取操作,既對溫水提取液中雜質進行了篩除,同時二氯甲烷的沸點遠低于目標物,萃取液沸程較短,因此胺鮮酯損失較小,回收率較高。
采用UPLC-MS/MS對樣品的添加過程的準確性進行了輔助驗證,證實樣品的添加誤差可控制在±5%范圍內。
2.3 基質效應
基質效應是指基質成分和目標化合物在進行離子化時相互競爭而導致目標化合物信號強度有不同程度的增強或減弱的現象,包括基質增強效應和基質抑制效應[14]。基質效應通常采用基質匹配標準溶液曲線與溶劑標準溶液曲線的斜率之比進行評價,比值小于1,存在基質抑制效應,反之存在基質增強效應。基質效應會影響某些待測物定量與定性的準確性,因此要對基質效應進行考察評估,并采取有效措施進行消除或補償。通過基質匹配標準溶液曲線方程計算未知樣品的濃度,能夠有效消除基質效應對樣品真實濃度的影響[15]。
使用1.5.1節所述前處理方式處理空白樣品,制備基質提取液,配制相應的基質標準溶液,供儀器檢測。結果表明,通過UPLC-MS/MS檢測,胺鮮酯在蘋果基質中存在基質抑制效應;通過GC-MS檢測,存在基質增強效應(見表1)。因此,當采用不同的儀器檢測時,應采用相應的基質匹配標準曲線計算未知樣品濃度。
y: peak area;x: mass concentration, mg/L.
2.4 線性關系和定量限
使用GC-MS分析基質匹配標準溶液,在0.005~0.2 mg/L范圍內,胺鮮酯的峰面積(y)與對應的質量濃度(x, mg/L)呈現良好的線性關系,相關系數大于0.999(見表1)。
以3倍信噪比(S/N)確定方法的檢出限(LOD),以最低添加、能準確定量的濃度確定方法的定量限(LOQ)。蘋果中胺鮮酯的LOD和LOQ分別為0.002 4 mg/kg和0.008 mg/kg。
2.5 回收率和精密度
按1.5節描述向空白基質中分別加入低、中、高3個水平的混合標準溶液,進行加標回收試驗(n=3)。結果表明,使用GC-MS測定時,蘋果中胺鮮酯的平均加標回收率為74.1%~84.2%,相對標準偏差為1.5%~4.1%(見表2)。

表2 蘋果中胺鮮酯的平均回收率和精密度(n=3)
3 結論
本工作建立了QuEChERS結合反萃取的前處理技術,并結合GC-MS檢測蘋果中胺鮮酯的分析方法。該方法針對性強,快速、高效,靈敏度、精密度均較高,可為該農藥殘留限量的制定提供參考。在后續的工作中,應在合理的基質效應范圍內,擴展可應用的檢測儀器,進一步提高該農藥殘留檢測的靈敏度。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
意林原創版(2016年10期)2016-11-25 10:28:30
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12