PRiME HLB固相萃取/高效液相色譜-串聯質譜法同時快速測定雞蛋中48種獸藥殘留
2019-11-28 07:06:22
食品工業科技 2019年22期
(廣西-東盟食品藥品安全檢驗檢測中心,廣西南寧 530021)
為了預防和治療禽類各種疾病、促進禽類生長、降低死亡率,獸藥被廣泛用于禽類及其產品。但由于在獸藥使用過程中濫用、誤用以及不遵守休藥期等現象屢禁不止,造成獸藥藥物及其代謝產物在禽蛋中殘留,超標的現象時有發生,嚴重影響公眾的健康[1-2]。中華人民共和國農業部公告第235號明確規定了食品動物禁用的一系列藥物,并制定了這類藥物最高殘留限量(MRLs)[3],其中激素類、氯霉素類、喹諾酮類、磺胺類、硝基咪唑類、大環內酯類是禁限用藥物;喹乙醇代謝物、吩噻嗪類、苯二氮卓類、四環素類、吡啶類、青霉素類、頭孢類獸藥是禽類常用且容易濫用的藥物[4]。因此,建立一種快速、簡便、靈敏、可靠、可同時檢測食品中多類獸藥殘留的檢測方法十分必要。
液相色譜-串聯質譜法因具有高的靈敏度、選擇性以及分離能力,已成為同時檢測復雜動物組織樣品中多類獸藥殘留極其重要的分析方法[5-6]。但這些方法只能同時檢測獸藥中的2~4類,同時快速測定上述14類藥物在雞蛋中殘留量檢測方法報道甚少。在我國農業部235號公告對禽蛋制定的18種殘留限量中,僅制定了禽蛋中哌嗪、樂泰菌素、林可霉素、大觀霉素4種獸藥的檢測標準。磺胺類物質按《畜禽肉中十六種磺胺類藥物殘留量的測定 液相色譜-串聯質譜法》(GB/T 20759-2006)檢測,而此標準僅適用于牛肉、羊肉、豬肉、雞肉和兔肉,并未提及禽蛋[7]。《綠色食品 蛋與蛋制品》(NY/T 754-2011)中要求四環素、金霉素和土霉素按《動物源性食品中四環素類獸藥殘留量檢測方法 液相色譜-質譜/質譜法與高效液相色譜法》(GB/T 21317-2011)測定,而此標準僅適用于動物肌肉、內臟組織、水產品、牛奶,也未提及禽蛋。我國對于禽蛋中獸藥殘留的檢測相關報道甚少,只涉及到氯霉素、甲硝唑、硝基呋喃類和喹諾酮類藥物[8-9]。由此可看出我國禽蛋中獸藥殘留限量標準尚不全面,部分蛋禽養殖過程普遍使用的獸藥沒有制定殘留限量標準,部分獸藥殘留限量值的設定仍需探究;同時制定殘留限量標準的獸藥缺乏相應的檢測方法[10-11]。因此,開發建立禽蛋中獸藥殘留的檢測方法,考究禽蛋中獸藥殘留的種類,加強我國禽蛋獸藥殘留的風險評估,對于食品安全國家標準中獸藥最大殘留限量及相關檢測標準的制修訂工作具有重要意義,為食品安全監管工作提供重要的技術支撐[12]。
前處理采用PRiME HLB一步法[13-14]提取和凈化雞蛋樣品,前處理簡單,并且基質干擾較小、靈敏度較高,能得到較為滿意的效果。Oasis PRiME HLB固相小柱是在Oasis HLB固相小柱技術基礎上開發的新一代反相固相萃取吸附劑,可以有效地吸附樣品中蛋白質和磷脂類物質,較以往固相萃取劑具有更簡單、更快速、更潔凈的優勢[15-16]。因此本研究采用PRiME HLB固相萃取前處理樣品,結合液相色譜-串聯質譜儀檢測,建立了同時測定雞蛋中磺胺及其增效劑類(10種)、喹諾酮類(9種)、硝基咪唑類(5種)、喹乙醇代謝物(1種)、氯霉素類(3種)、吩噻嗪類(1種)、苯二氮卓類(1種)、大環內酯類(5種)、四環素類(4種)、吡啶類(1種)、青霉素類(4種)、頭孢類(2種)、激素類(1種)、抗病毒類(1種)共14類48種獸藥殘留的新方法,為禽類及其產品中殘留獸藥的分析提供借鑒。
1 材料與方法
1.1 材料與儀器
雞蛋 均來自廣西多個地市(包括南寧市、桂林市、柳州市、百色市等11個地區)共82批,所有雞蛋樣品均取可食用部分,絞碎均質后,-20 ℃冷凍保存,待檢測;磺胺甲基嘧啶、磺胺甲惡唑、磺胺二甲嘧啶、磺胺地索辛、磺胺間甲氧嘧啶、磺胺喹噁啉、磺胺嘧啶、磺胺鄰二甲氧嘧啶、磺胺氯噠嗪、甲氧芐啶、恩諾沙星、環丙沙星、沙拉沙星、洛美沙星、諾氟沙星、培氟沙星、氧氟沙星、達氟沙星、司帕沙星、地美硝唑、甲硝唑、洛硝噠唑、羥基甲硝唑、羥甲基甲硝咪唑、3-甲基噁喹啉-2-羧酸、氯丙嗪、地西泮、林可霉素、替米考星、紅霉素、阿奇霉素、羅紅霉素、四環素、土霉素、金霉素、強力霉素、氯羥吡啶、阿莫西林、氨芐青霉素、芐青霉素、鄰氯青霉素、頭孢氨芐、頭孢唑啉、地塞米松、金剛烷胺、氯霉素、氟苯尼考、甲砜霉素標準品 純度均大于90.0%,德國Dr.Ehrenstorfer公司;乙腈 色譜純,德國默克公司;乙酸銨 色譜純,美國Tedia公司;甲酸 分析純,廣東省化學試劑工程技術研究開發中心;所用水 超純水。
PRime HLB固相萃取小柱(6cc,500 mg)、MCX陽離子交換柱(6cc,150 mg) 美國Waters公司;QTRQ AP4500串聯四級桿液質聯用儀 美國AB公司;Milli-Q Advantage A10超純水機 德國Millipore公司;PGC753e型電子天平 艾德姆衡器(武漢)有限公司;Multi Reax型全自動振蕩器 Heidolph公司;X3R型高速冷凍離心機 Thermo公司;Genius NM32LA型氮氣發生器 PEAK公司;ATR Autovap S60型多樣品自動濃縮儀 力德生物科技(上海)有限公司。
1.2 實驗方法
1.2.1 樣品處理 稱取樣品約2.50 g(精確到0.01 g)于50 mL聚酯離心管中,用90%乙腈水溶液定容至10.0 mL,劇烈振蕩10 min后,10000 r/min冷凍離心5 min。取上清液約5 mL,加入固相小柱內(Oasis PRiME HLB,500 mg,6cc,無需活化),保持1 s/滴,收集流出液到4.0 mL,40 ℃氮吹至小于0.5 mL,用10%乙腈水溶液定容至1.0 mL,渦旋2 min,10000 r/min離心5 min,取上清液,過0.22 μm濾膜,即可[17]。
1.2.2 標準溶液的制備 分別準確稱取48種獸藥標準物質約10 mg,用乙腈溶解并定容至10 mL,制成1 g/L的單標儲備液。分別吸取0.1 mL至同一100 mL容量瓶中,用乙腈稀釋至刻度,制成1 μg/mL的混合標準溶液。
稱取2.50 g雞蛋樣品空白基質,分別加入混合標準溶液按1.2.1方法處理樣品,制得質量濃度分別為0.5、1、5、10、20、50 μg/kg的基質混合標準工作溶液。以基質混合標準工作溶液中48種獸藥的質量濃度為橫坐標,對應定量離子的色譜峰面積為縱坐標繪制標準曲線[18]。
1.2.3 色譜條件 正離子模式液相色譜條件:色譜柱CAPCELL PAK C18 MGⅢ-H(3 μm,2.0×100 mm),流動相A為0.1%甲酸水、B為0.05%甲酸乙腈。梯度洗脫程序如表1所示,流速0.3 mL/min,進樣體積20 μL,色譜柱溫度35 ℃。

表1 正離子梯度洗脫程序Table 1 Cation gradient elution procedure
負離子模式液相色譜條件:色譜柱ACQUITY UPLC? BEH C18(1.8 μm,2.1×50 mm),流動相C為5 mmol/L乙酸銨、流動相D為乙腈。梯度洗脫程序如表1所示:正離子的流速0.3 mL/min,進樣體積20 μL,色譜柱溫度45 ℃。負離子的流速0.3 mL/min,進樣體積20 μL,色譜柱溫度35 ℃。

表2 負離子梯度洗脫程序Table 2 Anion gradient elution procedure
1.2.4 質譜條件 離子源:電噴霧離子源(ESI),多反應監測(MRM);正離子掃描模式:電噴霧電壓(IS)5500 V;離子源溫度(TEM)550 ℃;氣簾氣(CUR)流速35 L/min;霧化氣(GS1)流速55 L/min;輔助加熱氣(GS2)流速55 L/min;射入電壓(EP)10 V。負離子掃描模式:電噴霧電壓(IS)-4500 V;離子源溫度(TEM)550 ℃;氣簾氣(CUR)流速35 L/min;霧化氣(GS1)流速50L/min;輔助加熱氣(GS2)流速50 L/min;射入電壓(EP)-10 V。
1.3 數據處理
使用MultiQuant 3.0.2分析軟件對數據進行定量定性分析。
2 結果與分析
2.1 質譜離子源參數的優化
本研究測定的目標化合物種類較多,極性相差大,在質譜中的響應差別也較大,有必要對質譜中信號響應較小的化合物進行離子源參數進行優化。分別選擇羥甲基甲硝咪唑、紅霉素、甲砜霉素對正、負離子源條件進行優化。將流動相與標準溶液注射泵鏈接,同時將流動相與標準溶液注入質譜儀中,分別調節質譜的電噴霧電壓(IS)、離子源溫度(TEM)、氣簾氣(CUR)流速、霧化氣(GS1)流速、輔助加熱氣(GS2)流速[19-22]。得到化合物響應最大時的離子源條件見1.2.4,48種獸藥的相關質譜參數見表3。
2.2 色譜條件的優化
2.2.1 色譜柱的選擇 分別對CAPCELL PAK C18 MGⅢ-H(3 μm,2.0×100 mm)、Waters ACQUITY UPLC? BEH C18(1.7 μm,2.0×150 mm)、Agilent ZORBAX Eclipse Plus C18(1.8 μm,2.1×150 mm)、Phenomenex Kinetex XB-C18(1.7 μm,2.1×100 mm)4種不同型號色譜柱對相同濃度的標準溶液掃描分析,考察其保留時間和分離效果[23-24]。結果發現四根柱子中CAPCELL PAK C18 MGⅢ-H(3 μm,2.0×100 mm)測定正離子項目組分保留時間明顯縮短,分離度更好,而ACQUITY UPLC? BEH C18(1.7 μm,2.0×150 mm)測定負離子項目組分的峰型更尖銳,靈敏度更高,因此選用上述兩種色譜柱分別對正離子和負離子進行測定。
2.2.2 流動相的選擇 在流動相系統選擇中,為了使目標物和干擾物達到分離,分別使用甲醇-水、乙腈-水、乙腈-0.1%甲酸水、乙腈-5 mmol/L乙酸銨水溶液(負離子)等多種流動相系統進行試驗,以采用乙腈-0.1%甲酸水溶液檢測正離子質譜的分離度和響應值最高,而采用乙腈-5 mmol/L乙酸銨水溶液檢測負離子質譜的分離度和響應值較高。所以采用多反應監測模式分開對正、負離子進行測定,使離子對試劑在質譜檢測器中響應提高。梯度洗脫時,通過改變流動相的比例,使48種殘留藥物都分離出來,待測組分保留時間重現性好,靈敏度高。
2.3 前處理條件的優化
2.3.1 提取溶劑的選擇 雞蛋中含有豐富的蛋白質和脂肪,因此在提取過程中,需要考慮沉淀蛋白和去除脂肪[25]。常見的提取劑為乙腈,乙腈同時還具有沉淀蛋白的作用,所以選擇乙腈作為提取溶劑[26]。本研究比較了70%乙腈、80%乙腈、90%乙腈、100%乙腈、含1%甲酸乙腈溶液5種溶劑,發現70%乙腈、80%乙腈在過柱時容易堵塞凈化柱,可能因為溶劑水分比較高,導致沉淀蛋白和去除脂肪能力減弱,同時水分含量高,在氮吹濃縮步驟時去除水分更為繁瑣費時;含1%甲酸乙腈可以較好的沉淀樣品中的蛋白質,但對一些堿性化合物如磺胺類回收率偏低;90%乙腈與100%乙腈比較,發現90%乙腈提取回收率較高,峰形較好,故采用90%乙腈進行提取,提取溶劑考察數據對比見圖1。

圖1 提取溶劑考察數據對比圖Fig.1 Comparison of extraction solvent investigation data

表3 48種獸藥的質譜參數Table 3 Mass spectrometric parameters of 48 veterinary drugs
2.3.2 固相萃取柱的選擇 樣品直接提取液含大量雜質如脂肪和磷脂類等物質,所以選擇經過固相萃取柱來盡可能凈化樣品溶液[27],本研究通過對waters HLB固相小柱和MCX陽離子交換固相萃取小柱進行加標回收比較,結果發現,HLB固相萃取過柱時比較容易堵塞凈化柱,回收率低,偏差較大,而使用陽離子交換柱的檢測響應值明顯偏低。PRiME HLB為新型固相HLB處理小柱,屬于過濾性除去雜質固相小柱,能有效的去除樣品中的蛋白質、磷脂類干擾物質,本研究比較了未使用PRiME HLB凈化(圖2)和使用PRiME HLB凈化(圖3)的樣品,在母離子質荷比為431和子離子質荷比為184條件下,對磷脂中的主要成分磷脂酰膽堿進行檢測,結果表明PRiME HLB小柱能去除大部分的磷脂。選用PRiME HLB前處理過程更加快速、高效、靈敏,能滿足雞蛋中48種藥物殘留的分析檢測要求,所以選用PRiME HLB進行凈化[28]。

圖2 未使用PRiME HLB凈化樣品的磷脂酰膽堿離子流圖Fig.2 Ion chromatogram of thepurified sample without PRiME HLB

圖3 使用PRiME HLB凈化后樣品磷脂酰膽堿離子流圖Fig.3 Ion Chromatogram of thepurified sample using PRiME HLB
2.3.3 定容溶劑的選擇 分別用10%乙腈、20%乙腈、30%乙腈、50%乙腈、100%乙腈定容上機考察,實驗發現使用100%乙腈、20%、30%、50%乙腈定容時多數目標物的峰型變寬,當用10%乙腈定容時峰型尖銳,因此選擇10%乙腈為定容溶劑。
2.4 線性范圍、檢出限和定量限
按1.2.2配制6個水平的基質匹配混合標準溶液,在選定的色譜條件和質譜參數下進行檢測。以目標化合物定量離子的峰面積(Y)為縱坐標、質量濃度(X,μg/L)為橫坐標繪制工作曲線,48種獸藥的線性范圍為0.5~50 μg/kg,相關系數r為0.9952~1.0000,線性良好。以定量離子信噪比S/N=3為樣品的檢出限,S/N=10為定量限,結果見表4。48種獸藥的檢出限為0.01~0.55 μg/kg,定量限為0.03~1.83 μg/kg。
2.5 回收率和精密度
在優化條件下采用空白雞蛋基質進行加標回收率及精密度試驗,樣品添加水平分別為1、5和50 μg/kg,分別對每個添加水平進行平行測試3次,計算平均回收率及相對標準偏差(RSD)(見表5)。各化合物的回收率為63.3%~111.4%,符合多殘留分析的要求。圖4、圖5為50 μg/kg 添加水平下,雞蛋中目標化合物的正、負離子流色譜圖。

圖4 雞蛋中45種獸藥的正離子流圖Fig.4 Positive ion flow diagram of 45 veterinary drugs in eggs

圖5 雞蛋中3種獸藥的負離子流圖Fig.5 Anion flow diagram of three veterinary drugs in eggs
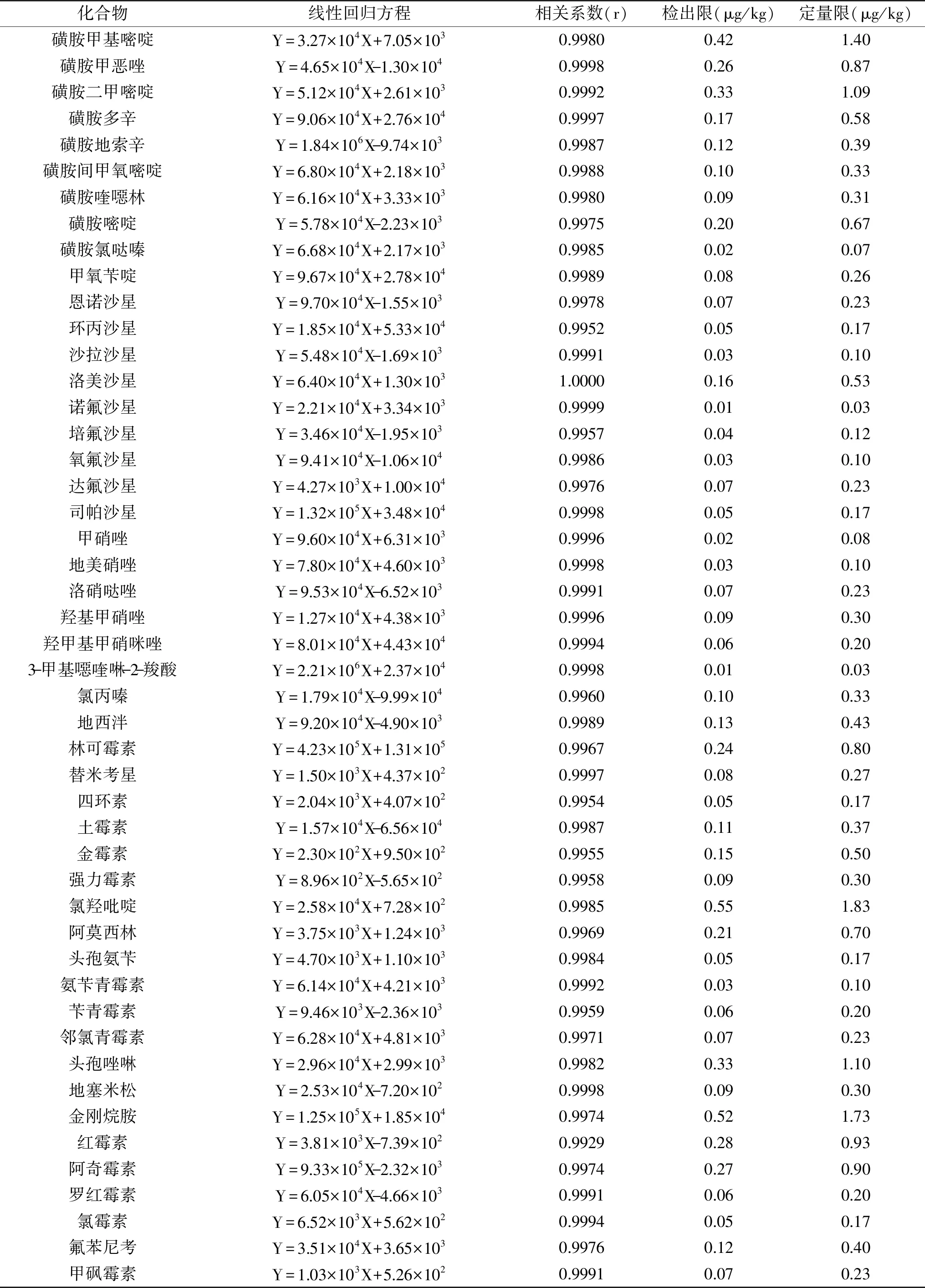
表4 雞蛋中48種獸藥殘留的線性方程、相關系數及檢出限、定量限Table 4 Linear equation,correlation coefficient,detection limit and quantitative limit of 48 veterinary drug residues in eggs
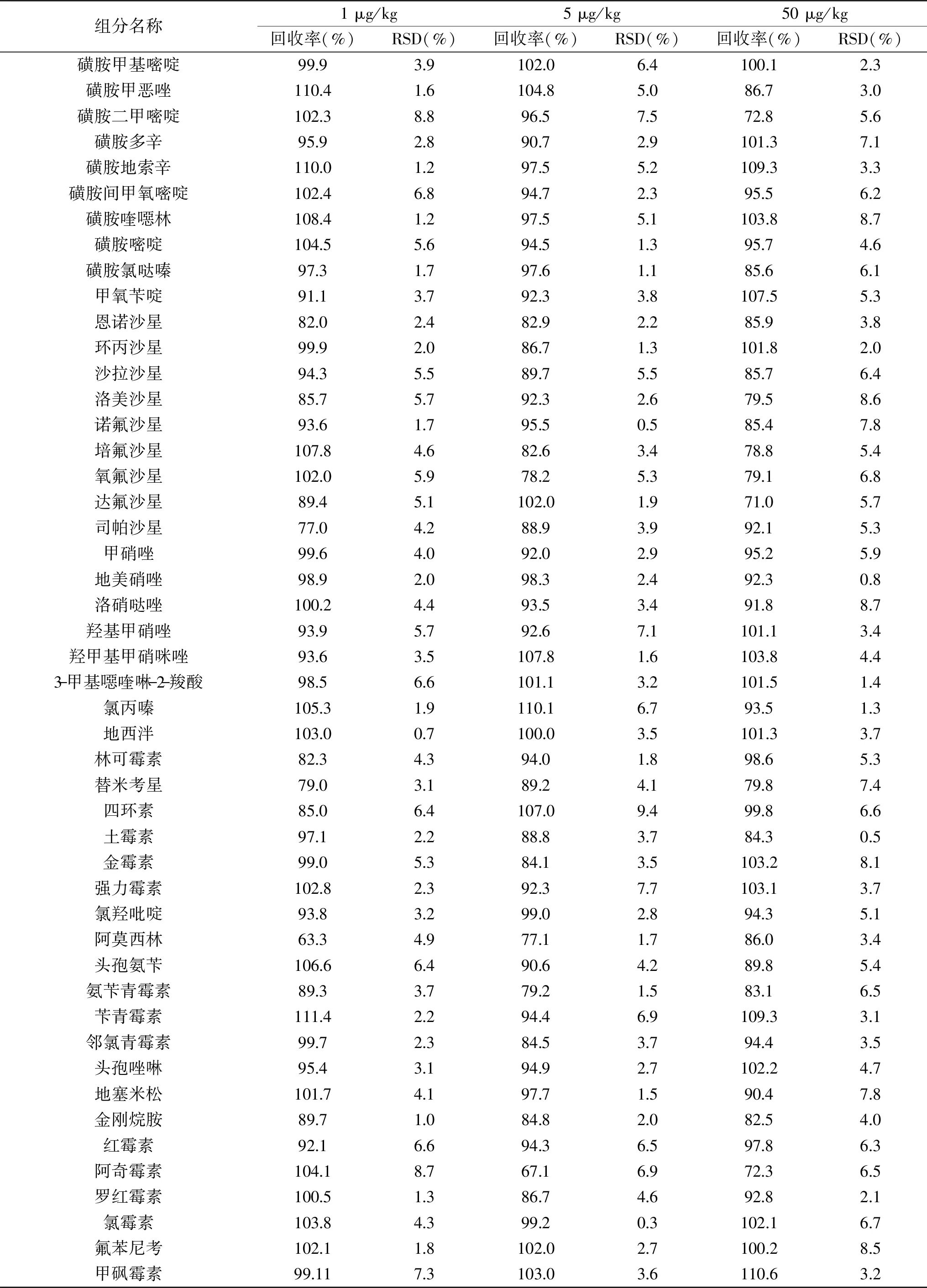
表5 雞蛋中48種獸藥殘留加標回收率及精密度(n=3)Table 5 The adding standard recovery rate and accuracy degree of 48 veterinary drugs in eggs(n=3)
2.6 實際樣品測定
采用本研究方法對廣西11個地市共82批次雞蛋進行獸藥殘留篩查,檢出10項目標化合物,針對檢出的10種化合物,采用法定標準GB/T 21316-2007、GB/T 21312-2007、GB/T 21318-2007、SN/T 1777.2-2007、GB 29699-2013、SN/T 4253-2015、GB/T 22338-2008對檢出項目進行測定,對本研究方法測得結果與標準方法測得結果進行比較,發現本研究方法測得結果與標準方法測得結果基本一致,表明本研究方法數據準確、可靠。結果見表6。
本文采用改進的Oasis PRiME HLB方法,建立了LC-MS/MS同時測定雞蛋中磺胺類及其增效劑、喹諾酮類、硝基咪唑類、喹乙醇代謝物、氯霉素類等14類48種獸藥殘留分析方法。與現行國家標準及行業標準相比,本方法可以同時分析多類獸藥殘留,簡單快速,縮短了檢驗周期,節約了檢驗的成本。本方法操作簡單,凈化效果好,靈敏度、準確度和精密度符合多獸殘檢測技術的要求,為各食品檢測機構提供簡單快速的方法來應對大批量的雞蛋樣品中禁限用獸藥的日常監管,為保證人民飲食安全提供了技術支持。
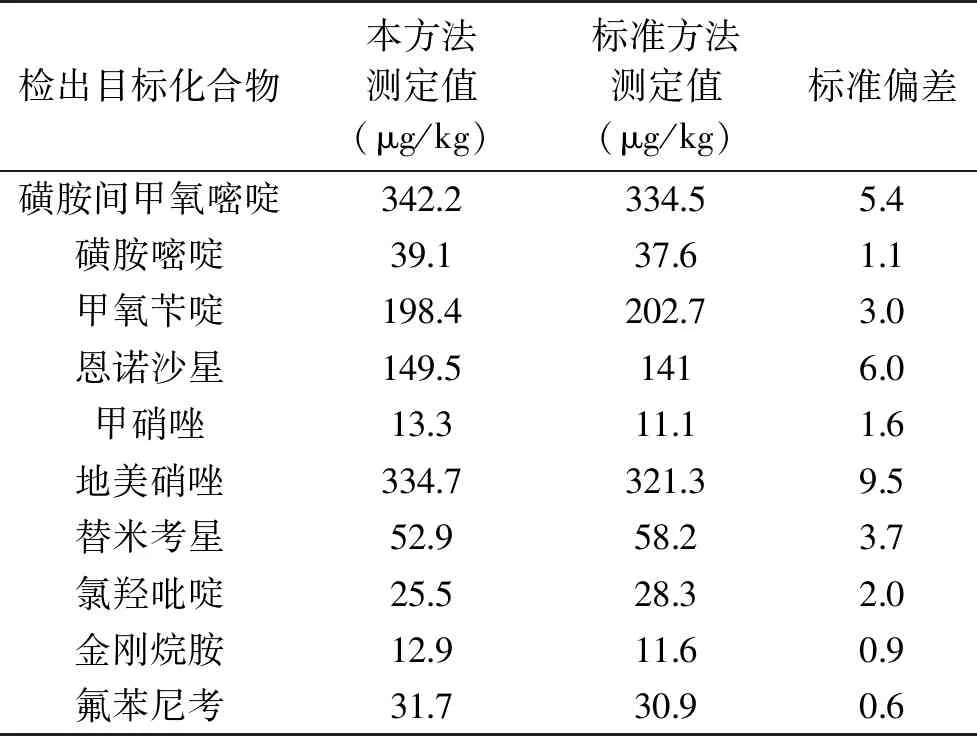
表6 本方法與標準方法測得值比較Table 6 Comparison of measured valuesbetween the test and the GB method
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
