CRISPR/Cas系統(tǒng)在活細(xì)胞染色體成像中的應(yīng)用
2020-02-14 15:44:33袁曦陳群何倩于冬梅秦培武
天津農(nóng)業(yè)科學(xué) 2020年1期
袁 曦 陳 群 何 倩 于冬梅 秦培武
摘? ? 要:CRISPR/Cas(Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated Proteins)是細(xì)菌抵抗外來入侵的一種自適應(yīng)免疫機(jī)制,利用CRISPR/Cas9系統(tǒng)可實(shí)現(xiàn)雙鏈DNA的剪切并誘導(dǎo)宿主細(xì)胞DNA修復(fù)機(jī)制,從而達(dá)到靶向編輯基因的目的。核酸酶失活的Cas9(Nuclease-deactivated Cas9,dCas9)耦聯(lián)效應(yīng)分子可以調(diào)控靶標(biāo)結(jié)合位點(diǎn)附近基因的表達(dá)、表觀遺傳修飾及特異染色體區(qū)域標(biāo)記。目前已開發(fā)出多種CRISPR/Cas9系統(tǒng),可對(duì)活細(xì)胞中重復(fù)或低重復(fù)序列基因位點(diǎn)進(jìn)行實(shí)時(shí)多位點(diǎn)同步成像,廣泛應(yīng)用于動(dòng)物和植物細(xì)胞中。基于CRISPR/Cas系統(tǒng)的活細(xì)胞染色體成像技術(shù)為研究活細(xì)胞染色體動(dòng)力學(xué)和三維染色體結(jié)構(gòu)提供了全新角度。本研究針對(duì)CRISPR/Cas系統(tǒng)的生源機(jī)制及其在活細(xì)胞成像的應(yīng)用和發(fā)展現(xiàn)狀進(jìn)行概述,以期為該領(lǐng)域的相關(guān)研究提供參考。
關(guān)鍵詞:CRISPR/Cas系統(tǒng);活細(xì)胞;染色體;成像
中圖分類號(hào):Q789? ? ? ? ?文獻(xiàn)標(biāo)識(shí)碼:A? ? ? ? ? ?DOI 編碼:10.3969/j.issn.1006-6500.2020.01.002
Application of CRISPR/Cas9 System in Living Cell Imaging
YUAN Xi, CHEN Qun, HE Qian, YU Dongmei, QIN Peiwu
(Center of Precision Medicine and Healthcare, Tsinghua-Berkeley Shenzhen Institute, Shenzhen, Guangdong 510855, China)
Abstract: CRISPR/Cas is a bacterial adaptive immune system. By cleaving specific strands of DNA and inducing DNA repair mechanism in host cells through CRISPR/Cas9 system, genome editing can be achieved. Nuclease-deactivated Cas9 (dCas9) can regulate gene expression, epigenetic modification, and label specific chromosomal regions via coupling effectors. Diverse CRISPR/Cas9 systems can target either the repetitive or low repetitive chromosome loci for live cell visualization, which is applicable to both animal and plant cells. Live cell chromosome imaging based on CRISPR/Cas system provides a new perspective for studying chromosome dynamics and three-dimensional genome structure. This review summarized the recent progress on the study of the molecular mechanism of CRISPR/Cas system and its application in living cell imaging.
Key words: CRISPR/Cas system; living cell; chromatin; imaging
1 CRISPR/Cas技術(shù)的作用原理
CRISPR(Clustered Regularly Interspaced Short Palindromic Repeats)是自然界中細(xì)菌抵御病毒侵染而進(jìn)化產(chǎn)生的一種天然免疫系統(tǒng),自2012年起被廣泛應(yīng)用于基因編輯研究中[1]。CRISPR/Cas(CRISPR-associated Proteins)系統(tǒng)在微生物中具備豐富的遺傳多樣性,已經(jīng)發(fā)現(xiàn)的CRISPR/Cas系統(tǒng)可分為兩大類,包括6種主要類型和33種亞型 [2]。每種CRISPR-Cas系統(tǒng)的組成結(jié)構(gòu)各具特點(diǎn),當(dāng)前應(yīng)用最廣泛的是化膿性鏈球菌Cas9(Streptococcus pyogenes Cas9)[3]。近年來,CRISPR/Cas系統(tǒng)在靶向基因編輯、基因激活和抑制、表觀遺傳修飾、全基因組篩選、RNA病毒檢測(cè)和活細(xì)胞染色體成像等領(lǐng)域取得了一系列突破性進(jìn)展[4-6]。
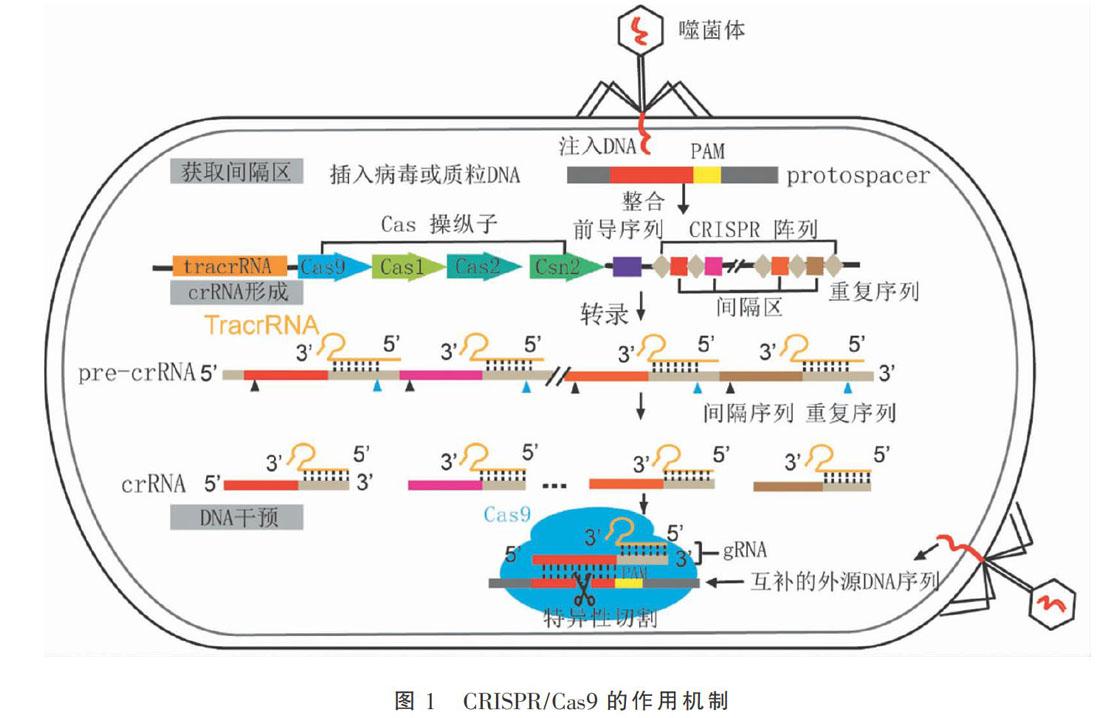
Cas9的免疫作用機(jī)制如圖1所示:細(xì)菌初次被來自噬菌體或病毒的外源DNA侵染時(shí),其CRISPR系統(tǒng)中的Cas1和Cas2酶識(shí)別并切割部分外源DNA中被稱為間隔區(qū)(Spacer)的DNA片段,并將其整合插入細(xì)菌DNA的回文重復(fù)序列(Palindromic Repeats)之間,從而獲取間隔區(qū);當(dāng)細(xì)菌再次被相同的外源病毒DNA侵染時(shí),宿主DNA會(huì)轉(zhuǎn)錄pre-crRNA(precursor-CRISPR RNA),隨后被切割成多個(gè)guide RNA(gRNA)。gRNA由crRNA(CRISPR RNA)和反式激活RNA(Trans-activating CRISPR RNA,TracrRNA)組成。crRNA中的spacer序列可與靶標(biāo)DNA的一條鏈堿基互補(bǔ)配對(duì),tracrRNA與crRNA的CRISPR重復(fù)區(qū)域(Repeats)堿基配對(duì)連接形成具有發(fā)卡結(jié)構(gòu)的gRNA,從而把這2個(gè)RNA融合形成單鏈的sgRNA(single guide RNA,sgRNA)。gRNA與Cas9結(jié)合后,crRNA搜索入侵病毒基因組DNA中能夠與其互補(bǔ)配對(duì)的位點(diǎn);Cas9識(shí)別PAM(Protospacer-Adjacent Motif)位點(diǎn)并錨定相應(yīng)區(qū)域,再通過Cas9核酸酶切割病毒DNA,從而實(shí)現(xiàn)阻礙病毒的入侵[7]。
CRISPR/Cas利用這種簡(jiǎn)單高效的識(shí)別系統(tǒng),精準(zhǔn)靶向某一特定DNA片段并進(jìn)行切割,切割后的雙鏈DNA在細(xì)胞內(nèi)可以通過兩種方式修復(fù),一種是非同源末端連接修復(fù)途徑(Non-Homologous End Joining Repair Pathway,NHEJ),NHEJ容易引起切割位點(diǎn)附近發(fā)生隨機(jī)插入或者刪除,從而破壞靶基因的正常表達(dá);另外一種是同源定向修復(fù)途徑(Homology Directed Repair Pathway,HDR),利用同源重組機(jī)制中可將目標(biāo)DNA片段整合到宿主基因組中。相比之前的基因編輯工具,如鋅指核酸酶(Zinc Finger Nuclease,ZFN)和轉(zhuǎn)錄激活因子樣效應(yīng)核酸酶(Transcription Activator-Like Effector Nuclease,TALEN),CRISPR/Cas9系統(tǒng)不需要進(jìn)行繁雜的人工蛋白改造,只需設(shè)計(jì)相應(yīng)的gRNA即可實(shí)現(xiàn)精準(zhǔn)定位并切割靶標(biāo)DNA,CRISPR/Cas基因組編輯系統(tǒng)的操作更加簡(jiǎn)單靈活、成本更低、特異性更高、識(shí)別范圍更廣[8]。
CRISPR核酸內(nèi)切酶的相關(guān)研究一直在快速發(fā)展,從較小核酸內(nèi)切酶到經(jīng)過修飾的核酸內(nèi)切酶,性質(zhì)各樣的核酸內(nèi)切酶使CRISPR系統(tǒng)基因組編輯功能更加強(qiáng)大。CRISPR/Cas9編輯基因是永久性的,對(duì)一些應(yīng)用而言并不理想。因此,研究者們對(duì)Cas9內(nèi)切酶核心區(qū)域進(jìn)行點(diǎn)突變,使dCas9(Nuclease-deactivated Cas9)喪失剪切DNA的內(nèi)切酶活力,但仍保留dCas9靶向特定DNA序列的能力[9]。利用dCas9與轉(zhuǎn)錄調(diào)節(jié)因子融合,能夠?qū)崿F(xiàn)可逆地調(diào)節(jié)基因表達(dá),在轉(zhuǎn)錄水平上激活或抑制非編碼RNA(Non-coding RNA, ncRNA)、天然反義轉(zhuǎn)錄產(chǎn)物(Natural Antisense Transcript, NAT)或MicroRNA的表達(dá)。下面分別介紹dCas9在轉(zhuǎn)錄調(diào)控、表觀遺傳修飾、細(xì)胞染色體成像方面的應(yīng)用概況。
2 CRISPR/dCas9在基因轉(zhuǎn)錄調(diào)控中的應(yīng)用
CRISPR/dCas9可通過與轉(zhuǎn)錄激活因子或者抑制因子融合實(shí)現(xiàn)對(duì)靶向基因的表達(dá)調(diào)控,其中dCas9-SAM(dCas9-Synergistic Activation Mediator)系統(tǒng)通過募集轉(zhuǎn)錄激活因子來調(diào)節(jié)靶基因表達(dá)。如dCas9與轉(zhuǎn)錄調(diào)控因子(如VP64和p65)融合,通過sgRNA靶向內(nèi)源啟動(dòng)子實(shí)現(xiàn)轉(zhuǎn)錄因子增強(qiáng)基因表達(dá)[10]。當(dāng)dCas9與轉(zhuǎn)錄抑制因子KRAB(Krüppel Associated Box)融合時(shí),募集組蛋白促進(jìn)異染色質(zhì)形成,可實(shí)現(xiàn)可逆抑制基因表達(dá)5~10倍。與依賴于細(xì)胞質(zhì)中mRNA降解來進(jìn)行基因抑制的RNA干擾機(jī)制不同,dCas9-KRAB能夠在DNA水平上進(jìn)行轉(zhuǎn)錄抑制,可用于ncRNAs、microRNAs、核定位RNA的轉(zhuǎn)錄抑制,擴(kuò)展和提高了非mRNA研究手段[11]。
表觀遺傳修飾是一系列針對(duì)遺傳物質(zhì)的精準(zhǔn)化學(xué)修飾,其可在不改變DNA序列的情況下導(dǎo)致特定基因選擇性沉默或活化。DNA甲基化及組蛋白修飾(例如甲基化、乙酰化)調(diào)節(jié)特定基因組區(qū)域是否被解壓縮且可被RNA聚合酶II接近,或者它是否被緊密地捆綁成轉(zhuǎn)錄沉默的異染色質(zhì)。表觀遺傳修飾作為基因表達(dá)調(diào)控的第二級(jí),是可遺傳和可逆的。CRISPR/dCas9技術(shù)的出現(xiàn)為表觀遺傳學(xué)研究提供新的途徑。將不同的效應(yīng)物連接到dCas9系統(tǒng)上,可實(shí)現(xiàn)表觀遺傳修飾改變,如組蛋白乙酰轉(zhuǎn)移酶p300與dCas9融合能夠使特異靶DNA序列附近的組蛋白乙酰化,從而激活基因表達(dá);而dCas9與去甲基化酶LSD1(Lysine-Specific Histone Demethylase 1A)融合能使靶序列附近的組蛋白去甲基化[12]。
3 CRISPR/dCas系統(tǒng)在活細(xì)胞染色體成像中的應(yīng)用
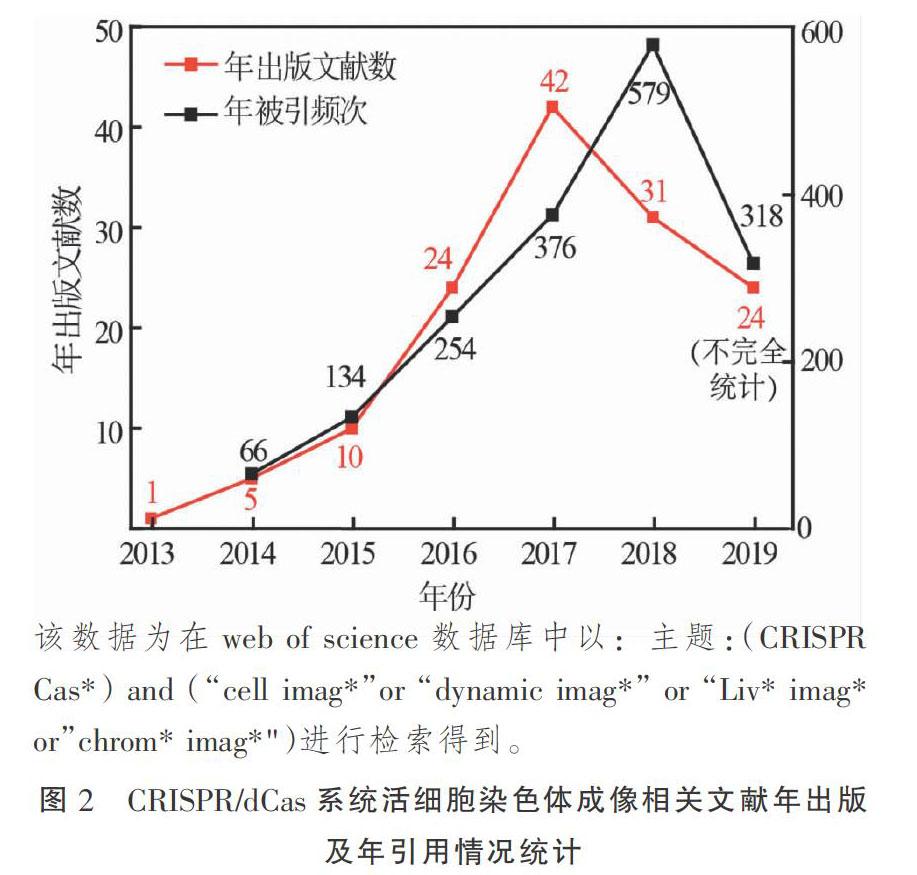
Chen等[13]于2013年首次報(bào)道了使用dCas9進(jìn)行染色體成像的相關(guān)研究,目前CRISPR/dCas系統(tǒng)已是活細(xì)胞染色體成像研究的一項(xiàng)重要工具。Web of Science數(shù)據(jù)庫檢索發(fā)現(xiàn),2016—2017年該主題文獻(xiàn)年出版數(shù)量快速增加,出版文獻(xiàn)年引用頻次也逐年遞增。這表明,近年來利用CRISPR/dCas系統(tǒng)進(jìn)行活細(xì)胞染色體成像逐漸得到了研究者們的密切
關(guān)注(圖2)。
dCas9與eGFP(enhanced Green Fluorescent Protein,eGFP)融合表達(dá)后,dCas9-eGFP復(fù)合體與sgRNA在細(xì)胞內(nèi)組裝成有功能的復(fù)合體,待復(fù)合體結(jié)合到靶標(biāo)DNA后,可通過熒光顯微鏡對(duì)活細(xì)胞中端粒等高重復(fù)染色體區(qū)域進(jìn)行動(dòng)態(tài)觀察。在非重復(fù)基因座上靶向36~73個(gè)sgRNA,該系統(tǒng)能夠觀測(cè)到MUC4基因座內(nèi)的非重復(fù)基因組區(qū)域。為實(shí)現(xiàn)多個(gè)gRNA的復(fù)用并將數(shù)十個(gè)不同的gRNAs高效遞送到單細(xì)胞中,Gu等 [14]擴(kuò)展了dCas9-eGFP成像方法,開發(fā)了一種稱為嵌合gRNA寡核苷酸陣列(Chimeric Array of gRNA Oligonucleotides,CARGO)的技術(shù)。CARGO技術(shù)將幾十個(gè)不同的gRNAs組成陣列遞送到單細(xì)胞中,從而精確識(shí)別獨(dú)特的非重復(fù)DNA片段,再利用多種熒光分子對(duì)其進(jìn)行標(biāo)記,實(shí)現(xiàn)了對(duì)特異DNA片段的動(dòng)態(tài)觀測(cè)。利用該技術(shù),Gu等[14]定量地測(cè)定了胚胎干細(xì)胞中發(fā)生分化相關(guān)活性的增強(qiáng)子和啟動(dòng)子的運(yùn)動(dòng)變化情況。
由于dCas9蛋白傾向定位在核仁中,dCas9-eGFP方法在核仁中會(huì)引發(fā)較高的背景信號(hào)。為了增強(qiáng)熒光信號(hào)強(qiáng)度和提升信噪比,可用更多的熒光蛋白(Fluorescent Protein,F(xiàn)P)分子標(biāo)記dCas9,例如通過使用超新星標(biāo)記系統(tǒng)(Sun-Tag),即利用一般對(duì)照不可誘導(dǎo)4號(hào)(General Control Non-Inducible 4,GCN4)肽序列與特異納米抗體之間相互作用募集多個(gè)FP[15]。利用dCas9-SunTag可以實(shí)現(xiàn)僅用20種不同的sgRNA就能連續(xù)跟蹤MUC4基因的非重復(fù)區(qū)域[16- 17]。
上述方法是針對(duì)改造過的dCas9與熒光蛋白融合進(jìn)行基因組成像,另外一種可替代方式是使用修飾的sgRNA對(duì)基因組位點(diǎn)進(jìn)行成像。這些經(jīng)修飾的sgRNA可以募集與FP融合的序列特異性RNA結(jié)合蛋白。例如經(jīng)修飾、包含多個(gè)重復(fù)RNA適配體的sgRNA,這些適配體可以特異性結(jié)合其同源結(jié)合蛋白(Cognate Binding Protein,CBP)。最廣泛使用的RNA適配體是MS2,一種源自噬菌體MS2的RNA莖環(huán)結(jié)構(gòu),其可以特異性結(jié)合MS2外殼蛋白(MS2 Coat Protein,MCP)。Qin等[18]的研究表明,基于MS2的系統(tǒng)可以用單個(gè)sgRNA對(duì)低重復(fù)序列位點(diǎn)進(jìn)行成像,增加MS2片段的重復(fù)次數(shù),可顯著提高信噪比和靈敏度。使用4個(gè)序列特異sgRNA,每個(gè)含有16個(gè)MS2適體,對(duì)低重復(fù)序列區(qū)域進(jìn)行標(biāo)記后,使用晶格光片顯微鏡(Lattice Light Sheet Microscopy,LLSM)可實(shí)現(xiàn)對(duì)細(xì)胞周期中天然染色質(zhì)基因座的跟蹤,并確定差異轉(zhuǎn)錄活性和非活性區(qū)域在細(xì)胞核中的定位。上述研究結(jié)果表明,MS2修飾的sgRNA方法可有效地監(jiān)測(cè)活細(xì)胞中重復(fù)和非重復(fù)基因組區(qū)域的位置和動(dòng)態(tài)。
此外,將CRISPR-dCas9和Pumilio RNA結(jié)合蛋白相結(jié)合建立的Casilio系統(tǒng),也可用于活細(xì)胞基因組位點(diǎn)的標(biāo)記[19]。Pumilio和Fem3 mRNA結(jié)合因子(Fem3 mRNA-Binding Factor,F(xiàn)BF)蛋白共享保守的Pumilio/FBF(PUF)RNA結(jié)合結(jié)構(gòu)域,該結(jié)構(gòu)域可通過設(shè)計(jì)用以結(jié)合特定的8聚體RNA序列(PUF-binding site,PBS)。Cheng等[19]通過設(shè)計(jì)sgTelomere和sgCentromere,使其分別攜帶多個(gè)PUF結(jié)合位點(diǎn),并將dCas9和Clover-PUF/sgCentromere-20×PBS及Ruby-PUF/sgTelomere-25×PBS在HEK293細(xì)胞內(nèi)共表達(dá),成功實(shí)現(xiàn)了在同一細(xì)胞內(nèi)同時(shí)對(duì)染色體端粒和中心粒進(jìn)行成像觀察。
4 CASFISH及CRISPR/dCas9與DNA FISH結(jié)合在染色體成像中的應(yīng)用
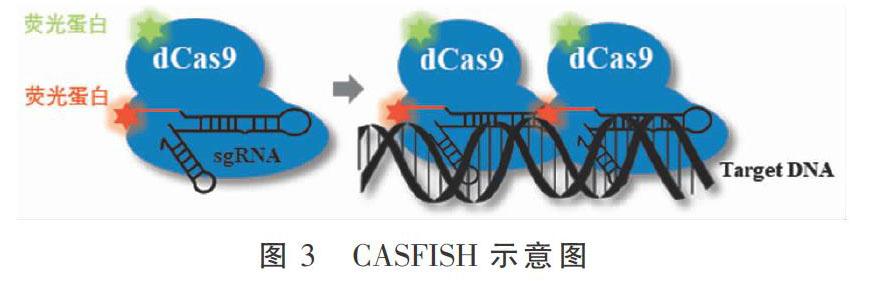
熒光原位雜交(Fluorescencein Situ Fybridization,
FISH)技術(shù)是利用與熒光基團(tuán)耦合的核苷酸作為探針,在細(xì)胞內(nèi)與相應(yīng)的靶DNA或RNA分子雜交,通過在熒光顯微鏡下觀察熒光信號(hào)而對(duì)組織、細(xì)胞或染色體上DNA或RNA進(jìn)行定性或定位分析的一種技術(shù)[20](圖3)。FISH迄今已發(fā)展了30多年,其在識(shí)別DNA和RNA序列方面具有高度敏感性和特異性,并且能在單細(xì)胞水平上同時(shí)對(duì)多個(gè)染色體位點(diǎn)進(jìn)行可視化追蹤[21],因此在解決單個(gè)染色體或整個(gè)基因組的結(jié)構(gòu)、突變和進(jìn)化等有關(guān)方面得到了廣泛應(yīng)用[22]。但由于DNA FISH需要經(jīng)過甲酰胺和熱處理使DNA變性以便于與熒光核酸探針進(jìn)行雜交,該過程可能會(huì)對(duì)生物體結(jié)構(gòu)和基因組織的完整性造成破壞,無法準(zhǔn)確的反映其原有空間結(jié)構(gòu)[23]。CASFISH(Cas9-mediated FISH)利用CRISPR/Cas9復(fù)合物具有識(shí)別特異靶DNA的特性來標(biāo)記序列特異性的染色體位點(diǎn)[24],DNA不需要經(jīng)過變性處理,有利于保留細(xì)胞形態(tài)和基因組結(jié)構(gòu)。CASFISH既可對(duì)含有高重復(fù)序列的染色體位點(diǎn)進(jìn)行標(biāo)記,也可通過使用多個(gè)sgRNA將Cas9-熒光蛋白復(fù)合物靶向目標(biāo)位點(diǎn),從而標(biāo)記非重復(fù)染色體位點(diǎn)。不同熒光標(biāo)記的dCas9/sgRNA還可對(duì)細(xì)胞中的多個(gè)染色體位點(diǎn)進(jìn)行同步多色標(biāo)記,以便于研究多個(gè)染色體位點(diǎn)之間的空間關(guān)系。由于CASFISH是利用dCas9介導(dǎo)的酶促反應(yīng)對(duì)特定序列進(jìn)行標(biāo)記,在優(yōu)化條件下15 min內(nèi)即可完成標(biāo)記,相對(duì)DNA FISH方法更加快捷[24]。將dCas9與Halo標(biāo)簽融合,形成的dCas-Halo可用于各種熒光染料標(biāo)記[24-25],再利用靶向不同DNA序列的sgRNA組合,可增加熒光染料選用的靈活性和多樣性,并降低開發(fā)靶向多重基因位點(diǎn)熒光探針的成本。
Takei等[26]和Guan等[27]提出了一種先跟蹤后識(shí)別(Track First and Identify Later)的策略,即將CRISPR/dCas系統(tǒng)和DNA Sequential FISH(seqFISH)相結(jié)合的方法來對(duì)活細(xì)胞內(nèi)的多個(gè)染色體位點(diǎn)進(jìn)行動(dòng)態(tài)追蹤(圖4)。首先用CRISPR/dCas9系統(tǒng)對(duì)多個(gè)染色體位點(diǎn)進(jìn)行單色實(shí)時(shí)成像。當(dāng)動(dòng)態(tài)記錄結(jié)束時(shí)再將細(xì)胞固定,連續(xù)多次使用DNA FISH來識(shí)別每個(gè)位點(diǎn)的身份。這一方法將染色體位點(diǎn)的動(dòng)態(tài)追蹤任務(wù)和這些位點(diǎn)的獨(dú)特識(shí)別分離,將CRISPR標(biāo)記和seqFISH兩者優(yōu)勢(shì)相結(jié)合,實(shí)現(xiàn)了在單一顏色通道下對(duì)活細(xì)胞的多個(gè)染色體位點(diǎn)的動(dòng)態(tài)追蹤。為了能夠在實(shí)時(shí)成像之后快速且連續(xù)的進(jìn)行DNA FISH操作,Guan等[27]優(yōu)化了FISH方法,使得整個(gè)染色過程能在1 min內(nèi)完成,極大地縮短了試驗(yàn)周期。每輪成像檢測(cè)后利用甲酰胺溶液洗滌以去除上一輪結(jié)合的DNA探針,避免連續(xù)多次FISH試驗(yàn)中DNA探針信號(hào)之間的干擾。利用該方法可同時(shí)對(duì)7個(gè)染色體位點(diǎn)進(jìn)行鑒定,DNA FISH經(jīng)過20多輪的染色和洗滌后,仍能保持高強(qiáng)度的熒光信號(hào)和高效率的清洗效果[27]。這種將實(shí)時(shí)和固定成像聯(lián)合使用的方法同樣可用于RNA-FISH中對(duì)多個(gè)靶標(biāo)RNA成像。
5 CRISPR/dCas系統(tǒng)在細(xì)胞染色體成像中的雙色標(biāo)記
在活細(xì)胞中對(duì)染色體進(jìn)行動(dòng)態(tài)成像往往需要能夠同時(shí)觀測(cè)一個(gè)以上的基因位點(diǎn),如研究表觀遺傳調(diào)控機(jī)制就涉及跟蹤兩個(gè)基因座染色質(zhì)的相互作用。目前CRISPR/dCas9系統(tǒng)通過以下兩種策略對(duì)活細(xì)胞中的基因組進(jìn)行雙色標(biāo)記。
一種策略是將來自不同菌種的dCas9蛋白耦合不同熒光蛋白[28-29](圖5A)。由于源自不同菌種的Cas9識(shí)別特定的PAMs序列且sgRNA骨架不同,彼此互不干擾,因此可以對(duì)不同基因組位點(diǎn)進(jìn)行多色標(biāo)記。除了化膿性鏈球菌(Streptococcus pyogenes,SP)Cas9之外,來自腦膜炎奈瑟氏球菌(Neisseria meningitides,Nm)、嗜熱鏈球菌(Streptococus thermophiles,St1)和金黃色葡萄球菌(Staphylococcus aureus,Sa)等的Cas9蛋白可以任意組合實(shí)現(xiàn)多色CRISPR成像。利用不同熒光染料標(biāo)記的dCas9-sgRNA可以確定不同染色體上基因座之間的核內(nèi)距離或同一染色體上兩個(gè)基因座之間的空間距離[29]。與SPCas9相比,NmCas9和St1Cas9能識(shí)別更長的PAM序列,因此其靶向設(shè)計(jì)受到了一定限制。此外,熒光標(biāo)記的dCas9蛋白體積較大,當(dāng)將這些融合蛋白引入同一個(gè)真核細(xì)胞時(shí),增加了轉(zhuǎn)染和病毒感染的難度。
另外一種策略是通過修飾sgRNA以實(shí)現(xiàn)基因組位點(diǎn)雙色成像(圖5B)。將RNA發(fā)夾結(jié)構(gòu)引入sgRNA的3'末端,使sgRNA轉(zhuǎn)化為支架RNA(scaffold RNA,scRNA),當(dāng)與dCas9共表達(dá)時(shí),可以將與熒光蛋白融合的RNA結(jié)合蛋白募集到靶點(diǎn)以實(shí)現(xiàn)特異標(biāo)記[30-32]。如Fu等[30]運(yùn)用此方法在sgRNA的3'末端引入了MS2和PP7,生成的sgRNA-MS2 和sgRNA-PP7分別與MCP-EGFP和mCherry-PCP融合蛋白相結(jié)合,實(shí)現(xiàn)了以不同顏色同時(shí)標(biāo)記主要和次要衛(wèi)星區(qū)域以及同時(shí)標(biāo)記小鼠染色體12上兩個(gè)單獨(dú)的基因位點(diǎn)。Shao等[32]的研究表明,此方法能夠長期同步對(duì)端粒和著絲粒的高重復(fù)區(qū)域進(jìn)行穩(wěn)定成像,并且基于sgRNA的標(biāo)記方法比基于Cas9的標(biāo)記方法更加耐受光漂白,這對(duì)于染色體動(dòng)態(tài)的、連續(xù)的和長期的跟蹤更具優(yōu)勢(shì)。此外,Zalatan等[33]基序可用于BFP融合的com蛋白識(shí)別,因此通過設(shè)計(jì)含有com基序的scRNA,可實(shí)現(xiàn)以不同顏色標(biāo)記第三個(gè)基因座。通過修飾sgRNA實(shí)現(xiàn)基因組位點(diǎn)雙色成像的最大的優(yōu)點(diǎn)在于,只使用一種dCas9蛋白即可實(shí)現(xiàn)染色體位點(diǎn)的多色標(biāo)記。由于SPCas9的PAM位點(diǎn)(NGG,N代表任何堿基),廣泛存在于基因組中,其靶向設(shè)計(jì)較NmCas9、St1Cas9等更具靈活性。與基于熒光蛋白修飾不同來源的Cas9的成像系統(tǒng)相比,修飾sgRNA進(jìn)行多色標(biāo)記更具靈活性,但此方法需要設(shè)計(jì)更多的融合蛋白,使得細(xì)胞系的構(gòu)建過程更加復(fù)雜。
6 CRISPR/dCas系統(tǒng)在細(xì)胞成像中的多色標(biāo)記技術(shù)
在染色體動(dòng)力學(xué)的研究中,往往需要同時(shí)對(duì)多個(gè)染色體位點(diǎn)進(jìn)行特異性和差異性標(biāo)記,但上述方法最多只能同時(shí)追蹤活細(xì)胞中的3個(gè)基因組位點(diǎn),難以滿足研究需要。為解決這一問題,Ma等[34]于2016年開發(fā)了一種名為CRISPRainbow的技術(shù)來實(shí)現(xiàn)對(duì)基因組多基因座進(jìn)行標(biāo)記,這是一種通過設(shè)計(jì)sgRNA以結(jié)合不同熒光蛋白對(duì)特定基因位點(diǎn)成像的技術(shù)(圖6)。首先將發(fā)夾結(jié)構(gòu)RNA(如MS2、PP7和boxB)連接到sgRNA的3'端或莖環(huán)上,這些特殊的發(fā)卡結(jié)構(gòu)分別能被融合了不同熒光蛋白(BFP、RFP和GFP)的天然結(jié)合蛋白(如MCP、PCP和N22)所識(shí)別。因此,當(dāng)sgRNA與兩個(gè)相同的熒光蛋白組合時(shí),會(huì)產(chǎn)生3種基色(藍(lán)色、紅色和綠色);當(dāng)sgRNA分別與兩個(gè)不同的熒光蛋白組合時(shí),3種基色兩兩疊加會(huì)產(chǎn)生青色(BFP+GFP)、品紅色(RFP+BFP)和黃色(GFP+RFP);而當(dāng)sgRNA同時(shí)與3種熒光蛋白組合時(shí),3種基色疊加將得到白色。雙重雙色標(biāo)記是通過sgRNA結(jié)合不同的熒光蛋白來實(shí)現(xiàn)的。這一技術(shù)能夠動(dòng)態(tài)地對(duì)多個(gè)不同的染色體位點(diǎn)進(jìn)行可視化觀察,目前已實(shí)現(xiàn)在活細(xì)胞中同時(shí)對(duì)6個(gè)染色體基因座同時(shí)成像[34]。與基于熒光蛋白修飾Cas9進(jìn)行多色標(biāo)記相比,CRISPRainbow只需利用SPCas9結(jié)合融合了不同熒光蛋白的sgRNA,標(biāo)記顏色的范圍更容易拓展。理論上,再增加一種顏色,CRISPRainbow將能夠同時(shí)對(duì)15個(gè)基因組位點(diǎn)進(jìn)行特異性的標(biāo)記。但由于在該系統(tǒng)中對(duì)sgRNA進(jìn)行了大量修飾,導(dǎo)致sgRNA的穩(wěn)定性不佳,而sgRNA的穩(wěn)定性是影響標(biāo)記效率的重要因素[35]。
為此,Ma等[36]提出了一種名為CRISPR sgRNA-Sirius的方法來改善CRISPR成像系統(tǒng)的標(biāo)記效率和靈敏度。首先,他們利用CRISPR-Broccoli系統(tǒng)[37]在活細(xì)胞中研究了在不同位點(diǎn)插入外源RNA對(duì)sgRNA穩(wěn)定性的影響,發(fā)現(xiàn)外源RNA插入sgRNA的tetraloop比3'-末端更穩(wěn)定[36]。然后通過進(jìn)一步優(yōu)化RNA適體的結(jié)構(gòu),設(shè)計(jì)出了3種CRISPR-Sirius結(jié)構(gòu)(sgRNA-Sirus-8XMS2、CRISPR sgRNA-Sirius-8XPP7和CRISPR sgRNA-Sirius-4X(MS2-PP7)。該結(jié)構(gòu)在保持靈活多色標(biāo)記的基礎(chǔ)上,提高了在活細(xì)胞中追蹤DNA的靈敏度,實(shí)現(xiàn)了同時(shí)可視化同一染色體上的多個(gè)不同基因座位,并測(cè)量了不同基因座的空間距離、觀測(cè)了其動(dòng)態(tài)行為。高效穩(wěn)定的多色標(biāo)記方法有望成為研究細(xì)胞周期進(jìn)程、表觀遺傳調(diào)控期間或細(xì)胞刺激反應(yīng)中染色體內(nèi)和染色體間結(jié)構(gòu)域動(dòng)態(tài)相互作用的有力工具。
7 CRISPR/dCas系統(tǒng)在植物細(xì)胞成像中的應(yīng)用
盡管CRISPR/dCas9系統(tǒng)于2013年就開始用于人體和哺乳動(dòng)物細(xì)胞特定基因組位點(diǎn)的可視化研究[13],但直到2017年Dreissig等[38]才首次將其應(yīng)用于植物細(xì)胞成像研究。在2017年以前,CRISPR/Cas9系統(tǒng)在植物細(xì)胞中主要用于基因編輯和轉(zhuǎn)錄調(diào)控研究。Dreissig等[38]利用分別標(biāo)記了eGFP和mRuby2的化膿性鏈球菌dCas9和金黃色葡萄球菌dCas9,觀測(cè)本塞姆氏煙草(Nicotiana benthamiana)葉細(xì)胞,開展蛋白質(zhì)-端粒相互作用動(dòng)力學(xué)研究,揭示了端粒在細(xì)胞間期的動(dòng)態(tài)運(yùn)動(dòng)規(guī)律。通過在葉片細(xì)胞中共同表達(dá)Sp-dCas9-mRuby、sgRNA-端粒和結(jié)合了GFP的端粒重復(fù)結(jié)合蛋白1(Telomeric Repeat Binding Protein 1,TRB1),研究者們還探討了染色體端粒的重復(fù)序列與TRB1結(jié)合比例之間的動(dòng)態(tài)關(guān)系。基于CRISPR/dCas9系統(tǒng)的細(xì)胞成像技術(shù)有望發(fā)展成為研究植物細(xì)胞染色體動(dòng)力學(xué)的重要技術(shù)。
8 展 望
盡管目前已開發(fā)出多種CRISPR/dCas9系統(tǒng)用于活細(xì)胞中重復(fù)或低重復(fù)序列基因組位點(diǎn)實(shí)時(shí)成像研究,但CRISPR/dCas9系統(tǒng)的靶向效率和成像靈敏度仍需進(jìn)一步提高。CRISPR/dCas9系統(tǒng)的靶點(diǎn)主要受PAM序列的限制,若想實(shí)現(xiàn)更多基因組位點(diǎn)的同步精確成像,需進(jìn)一步開發(fā)多樣化的Cas9系統(tǒng)與PAM識(shí)別位點(diǎn),從而進(jìn)一步擴(kuò)大CRISPR/dCas9系統(tǒng)的可識(shí)別靶點(diǎn)范圍。例如,尋找具有特異PAM位點(diǎn)的Cas9,或通過基因工程手段有目的性的設(shè)計(jì)Cas9,開發(fā)擁有新PAM特異性的Cas9突變體。SpCas9的PAM序列在動(dòng)植物基因組中廣泛存在,但其基因長度大于4 kb,增加了轉(zhuǎn)染和病毒感染的難度,研發(fā)小型的Cas9同源蛋白是發(fā)展其在細(xì)胞成像領(lǐng)域中應(yīng)用的重要方向。此外,優(yōu)化sgRNA的結(jié)構(gòu),設(shè)計(jì)更高效的sgRNA傳遞系統(tǒng),有助于充分發(fā)揮該技術(shù)標(biāo)記單拷貝基因的潛力。進(jìn)一步提高CRISPR信號(hào)或使用高靈敏度的尖端顯微鏡,可進(jìn)一步優(yōu)化信噪比,也有利于進(jìn)一步提高成像效率。高靈敏度和高效率的實(shí)時(shí)動(dòng)態(tài)成像技術(shù)有望幫助解決諸多基因組和染色質(zhì)領(lǐng)域研究的關(guān)鍵問題,CRISPR/dCas活細(xì)胞染色體成像技術(shù)的發(fā)展是解決這些問題的核心要義。
參考文獻(xiàn):
[1]JINEK M, CHYLINSKI K, FONFARA I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity[J]. Science,2012,337(6096):816-821.
[2]KOONIN E V, MAKAROVA K S, ZHANG F. Diversity, classification and evolution of CRISPR-Cas systems[J]. Current opinion microbiology,2017,37:67-78.
[3]SHMAKOV S, SMARGON A, SCOTT D, et al. Diversity and evolution of class 2 CRISPR-Cas systems[J]. Nature reviews microbiology,2017,15(3):169-182.
[4]KNOTT G J, DOUDNA J A. CRISPR-Cas guides the future of genetic engineering[J]. Science, 2018,361(6405):866-869.
[5]PRASHANT M, LUHAN Y, ESVELT K M, et al. RNA-guided human genome engineering via Cas9[J]. Science,2013,339(6121):823-826.
[6]HSU P D, LANDER E S, ZHANG F. Development and applications of CRISPR-Cas9 for genome engineering[J]. Cell,2014,157(6):1262-1278.
[7]JIANG F, DOUDNA J A. CRISPR-Cas9 Structures and Mechanisms[J].Annual review of biophysics,2017,46:505-529.
[8]GAJ T, GERSBACH C A, BARBAS C R. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering[J]. Trends in biotechnology,2013,31(7):397-405.
[9]QI L S, LARSON M H, GILBERT L A, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression[J]. Cell,2013,152(5):1173-1183.
[10]ZHANG Y, YIN C, ZHANG T, et al. CRISPR/gRNA-
directed synergistic activation mediator (SAM) induces specific, persistent and robust reactivation of the HIV-1 latent reservoirs[J]. Scientific reports,2015,5:16277.
[11]GILBERT L A, LARSON M H, MORSUT L, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes[J]. Cell,2013,154(2):442-451.
[12]SHRIMP J H, GROSE C, WIDMEYER S, et al. Chemical control of a CRISPR-Cas9 acetyltransferase[J]. ACS chemical biology,2018,13(2):455-460.
[13]CHEN B, GILBERT L A, CIMINI B A, et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system[J]. Cell,2013,155(7):1479-1491.
[14]GU B, SWIGUT T, SPENCLEY A, et al. Transcription-coupled changes in nuclear mobility of mammalian cis-regulatory?elements[J]. Science,2018,359(6379):1050-1055.
[15]TANENBAUM M E, GILBERT L A, LEI S Q, et al. A protein-tagging system for signal amplification in gene expression and fluorescence imaging[J]. Cell, 2014,159(3):635-646.
[16]WU X T, MAO S Q, YING Y C, et al. Progress and challenges for live-cell imaging of genomic loci using CRISPR-based platforms[J]. Genomics proteomics bioinformatics, 2019,17(2): 119-128.
[17]YE H, RONG Z, LIN Y. Live cell imaging of genomic loci using dCas9-SunTag system and a bright fluorescent protein[J]. Protein cell,2017,8(11):853-855.
[18]QIN P W, PARLAK M, KUSCU C, et al. Live cell imaging of low-and non-repetitive chromosome loci using CRISPR-Cas9[J]. Nature communications,2017,8:14725.
[19]CHENG A W, JILLETTE N, LEE P, et al.Casilio: a versatile CRISPR-Cas9-Pumilio hybrid for gene regulation and genomic labeling[J].Cell research, 2016,26(2):254-257.
[20]CUI C H, SHU W, LI P N. Fluorescence in situ hybridization: cell-based genetic diagnostic and research applications[J].Frontiers in cell and developmental biology, 2016,4:89.
[21]RIEGEL M. Human molecular cytogenetics: from cells to nucleotides[J].Genetics and molecular biology,2014,37(1):194-209.
[22]JIANG J. Fluorescence in situ hybridization in plants: recent developments and future applications[J]. Chromosome research,2019, 27(3):153-165.
[23]LEVSKY J M, SINGER R H. Fluorescence in situ hybridization: past, present and future[J]. Journal of cell science,2003,116(Pt 14):2833-2838.
[24]DENG W, SHI X, TJIAN R, et al. CASFISH: CRISPR/Cas9-mediated in situ labeling of genomic loci in fixed cells[J].Proceedings of the national academy of sciences of the united states of America, 2015,112(38):11870-11875.
[25]ENCELL L P, FRIEDMAN O R, ZIMMERMAN K, et al. Development of a dehalogenase-based protein fusion tag capable of rapid, selective and covalent attachment to customizable ligands[J].Current chemical genomics,2012,6:55-71.
[26]TAKEI Y, SHAH S, HARVEY S, et al. Multiplexed dynamic imaging of genomic loci by combined CRISPR imaging and DNA sequential FISH[J]. Biophysical journal,2017,112(9):1773-1776.
[27]GUAN J, LIU H, SHI X, et al. Tracking multiple genomic elements using correlative CRISPR imaging and sequential DNA FISH[J]. Biophysical journal,2017,112(6):1077-1084.
[28]CHEN B, HU J, ALMEIDA R, et al. Expanding the CRISPR imaging toolset with Staphylococcus aureus Cas9 for simultaneous imaging of multiple genomic loci[J]. Nucleic acids research,2016,44(8):75.
[29]MA H, NASERI A, REYES-GUTIERREZ P, et al. Multicolor CRISPR labeling of chromosomal loci in human cells[J]. Proceedings of the national academy of sciences of the united states of America, 2015,112(10):3002-3007.
[30]FU Y, ROCHA P P, LUO V M, et al. CRISPR-dCas9 and sgRNA scaffolds enable dual-colour live imaging of satellite sequences and repeat-enriched individual loci[J]. Nature communications,2016,7:11707.
[31]WANG S, SU J H, ZHANG F, et al. An RNA-aptamer-based two-color CRISPR labeling system[J]. Sciencereport,2016,6:26857.
[32]SHAO S, ZHANG W, HU H, et al. Long-term dual-color tracking of genomic loci by modified sgRNAs of the CRISPR/Cas9 system[J]. Nucleic acids research,2016,44(9):e86.
[33]ZALATAN J G, LEE M E, ALMEIDA R, et al. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds[J]. Cell,2015,160(1-2):339-350.
[34]MA H, TU L C, NASERI A, et al. Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow[J]. Nature biotechnology,2016,34(5):528-530.
[35]MA H, TU L C, NASERI A, et al. CRISPR-Cas9 nuclear dynamics and target recognition in living cells[J]. The Journal of cell biology,2016,214(5):529-537.
[36]MA H, TU L, NASERI A, et al. CRISPR-Sirius: RNA scaffolds for signal amplification in genome imaging[J]. Nature methods,2018, 5(11):928-931.
[37]FILONOV G S, MOON J D, SVENSEN N, et al. Broccoli: rapid selection of an RNA mimic of green fluorescent protein by fluorescence-based selection and directed evolution[J].Journal of the American chemical society,2014,136(46):16299-16308.
[38]DREISSIG S, SCHIML S, SCHINDELE P, et al. Live cell CRISPR-imaging in plants reveals dynamic telomere movements[J]. Plant journal,2017,91(4):565-573.
收稿日期:2019-11-21
基金項(xiàng)目:國家自然科學(xué)基金(31970752);Shenzhen Municipal Development and Reform Commission Subject Construction Project ([2017] 1434);Shenzhen Municipal Development and Reform Commission, Shenzhen Engineering Laboratory for Precision Medicine and Healthcare(SDRC[2015] 1950)
作者簡(jiǎn)介:袁曦 (1995—),女,重慶人,碩士,主要從事細(xì)胞成像方面的研究。
通訊作者簡(jiǎn)介:秦培武(1979—),男,黑龍江人,助理教授,博士生導(dǎo)師,主要從事染色體結(jié)構(gòu)與基因轉(zhuǎn)錄和細(xì)胞功能關(guān)系及熒光成像方法開發(fā)方面的研究工作。
