
利用Illumina MiSeq測序平臺分析沐川烏骨黑雞空腸微生物多樣性
2020-02-22 06:22:36廖娟王鋼喻世剛梁梓
江蘇農業科學 2020年24期
廖娟 王鋼 喻世剛 梁梓
摘要:為探索沐川烏骨雞腸道微生物多樣性,采用Illumina MiSeq測序平臺對10羽沐川烏骨雞的空腸內容物樣本中微生物的16S rDNA-V4變異區進行測序,利用UPARSE等軟件分析和統計樣品中的操作分類單元(OTUs)數量,并進行物種注釋及豐度分析,揭示樣品的物種構成。結果表明,共獲得667 466條tags,在97%相似水平上進行OTU分類,被歸類于3 154個OTUs,且由稀釋曲線可知此次測序結果較全面地覆蓋了沐川烏骨黑雞空腸微生物群落。門水平上,占優勢的門主要有厚壁菌門(Firmicutes)、擬桿菌門(Bacteroidetes)、變形菌門(Proteobacteria),但這3種優勢菌門在不同樣品中所占比例均有所差異。在屬水平上,在10個樣品中所占比例均大于1%的菌屬有乳桿菌屬(Lactobacillus)和Romboutsia菌屬。
關鍵詞:沐川烏骨黑雞;Illumina MiSeq測序;空腸;微生物;多樣性
中圖分類號: S831;S182 ?文獻標志碼: A ?文章編號:1002-1302(2020)24-0178-04
在動物的身體部位中,胃腸道中定殖著數量龐大、種類繁多的微生物,這類復雜的微生物群落被稱為腸道微生物[1]。腸道微生物菌群在宿主營養、發育、免疫及代謝等過程中起著重要作用[2-3]。近年來,腸道微生物多樣性成為很多學者的研究熱點,腸道微生物多樣性的研究方法較多,早期主要采用傳統的純培養方法[4],但是腸道中有很多厭氧型細菌,傳統純培養方法無法分離培養。隨著科學技術的發展,末端限制性片段長度多樣性分析、變性梯度凝膠電泳、溫度梯度凝膠電泳等技術廣泛運用于腸道微生物的分析中,這些方法雖能檢測一些厭氧型微生物,但這些研究技術獲得的信息覆蓋量仍不能充分反映出腸道微生物的多樣性。Illumina MiSeq技術因其方便、快速、準確率高和信息覆蓋量大等優點,被廣泛應用于兔[5]、人[6]、蜜蜂[7]等的腸道微生物研究中。林奕岑等[8]、徐帥等[9]采用Illumina MiSeq技術分別對普通雞的盲腸和回腸的微生物多樣性進行分析,但對沐川烏骨黑雞腸道微生物分析尚未見報道。沐川烏骨黑雞是四川省樂山市的優良地方品種資源,在沐川縣具有悠久的種源歷史,屬藥肉兼用型品種,因其全身烏黑而得名[10],其生產性能及健康程度與復雜的腸道微生物組成密切相關。本研究利用Illumina MiSeq測序平臺分析了沐川烏骨黑雞空腸微生物多樣性,旨在為相關研究提供理論依據。
1 材料與方法
1.1 試驗動物
試驗采用1日齡健康的沐川烏骨黑母雞,購于四川省樂山市沐川縣黑鳳凰烏骨雞業有限公司,采用相同的飼養和管理方式。
1.2 試驗方法
1.2.1 樣品采集 于30日齡時,隨機選取10羽健康個體,無菌操作采集每羽沐川烏骨黑雞的空腸內容物,依次編號K1~K10,并快速置于液氮罐中,帶回實驗室放入-80 ℃冰箱保存。
1.2.2 總DNA提取 采用SDS方法提取空腸內容物樣本基因組DNA,利用1%瓊脂糖凝膠電泳檢測DNA的純度和濃度,采用無菌水稀釋至1 ng/μL。
1.2.3 基因文庫構建及上機測序 以稀釋后的樣品基因組 DNA 為模板,使用16S rRNA-V4區特異性引物515F(5′-GTGCCAGCMGCCGCGGTAA-3′)和806R(5′-GGACTACHVGGGTWTCTAAT-3′)進行PCR擴增。PCR產物采用2%瓊脂糖凝膠進行電泳檢測后,切下目標條帶,用Thermo Scientific 公司GeneJET膠回收試劑盒進行回收。
使用建庫試劑盒(Thermofisher公司的Ion Plus Fragment Library Kit 48 rxns建庫試劑盒)進行文庫的構建,構建好的文庫經過定量、檢測合格后,使用Thermofisher的Ion S5TMXL進行上機測序。測序工作在諾禾致源生物信息有限公司完成。
1.3 生物信息學分析
測序完成后,原始數據使用Cutadapt對低質量部分進行剪切[11],再根據Barcode從得到的reads中拆分出各樣品數據,截去Barcode和引物序列得到的原始數據(raw reads)后,利用UCHIME Algorithm 軟件去除嵌合序列[12],得到有效數據(clean reads)。利用Uparse軟件對所有樣品的全部clean reads按97%的相似性進行操作分類單元(operational taxonomic unit,簡稱OTU)聚類。并對用Mothur方法與SILVA的SSUrRNA數據庫進行物種注釋分析[13-14]。采用Qiime軟件計算每個樣品的Alpha多樣性。
2 結果與分析
2.1 各樣品序列及OTUs信息
本試驗基于Illumina MiSeq測序平臺對10羽沐川烏骨黑雞的空腸內容物菌群V4區進行了測序,一共獲得667 466條tags,在97%的相似水平上進行OTU分類,被歸類于3 154個OTUs。其中,tags數量最高的是樣品K3,為86 261條;tags數量最低的是樣品K9,為47 679條。
2.2 Alpha多樣性分析
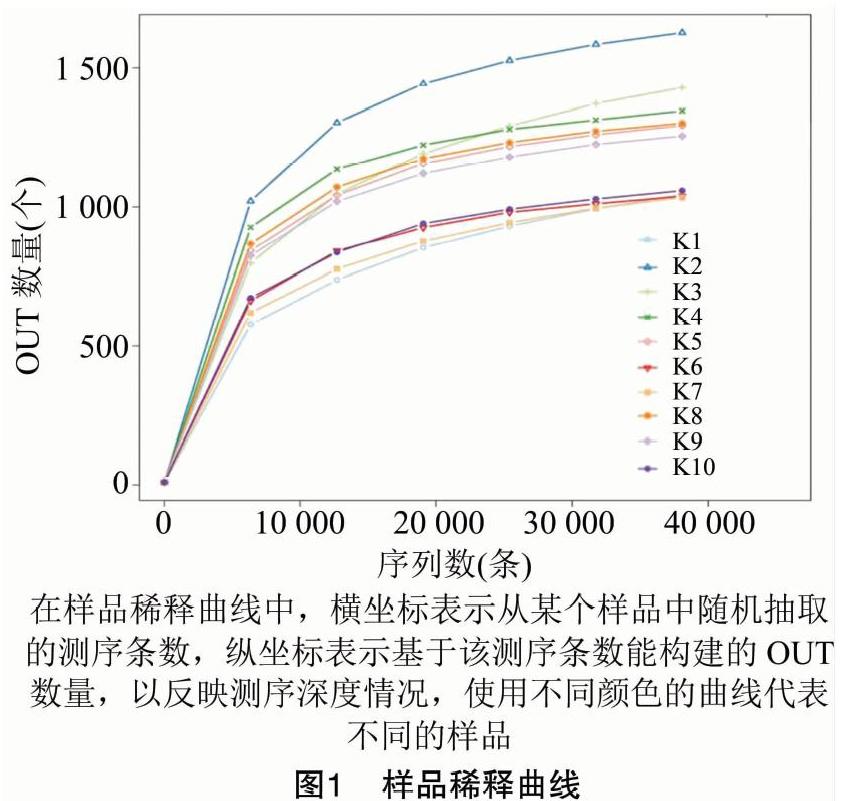
采用Alpha多樣性指數分析樣品內的微生物群落的豐富度和多樣性。從樣品稀釋曲線(圖1)可知,隨著測試深度的不斷加深,稀釋曲線逐漸趨于平坦,說明測序數據量漸進合理,10個樣本的覆蓋率(good's coverage)均達到了99.0%以上,表明更多的數據量僅會產生少量新OTUs,16S rRNA基因測序深度足以反映樣品的微生物多樣性。
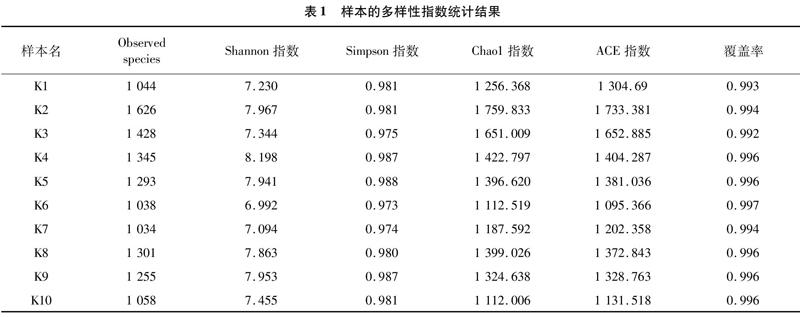
采用Chao1、ACE、Shannon、Simpson等指數表示,樣品中微生物群落的Alpha多樣性。Chao1或ACE指數越大,表明樣品群落豐富度越高;Shannon指數越高,表明群落物種多樣性越高;Simpson指數越高,表示其群落均勻度越高。由表1可知,樣品K2物種豐富度相對較高,樣品K4物種多樣性相對較高。
3.3 物種注釋
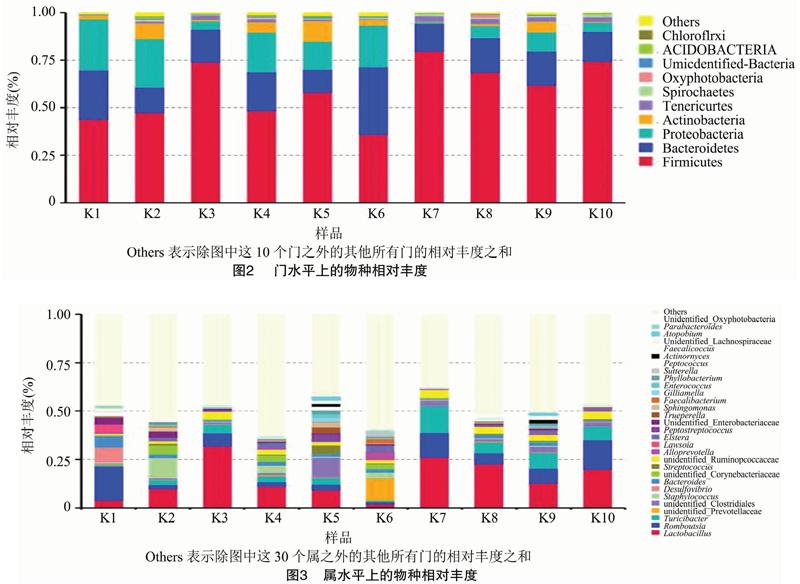
根據物種注釋結果,選取每個樣品在門水平上最大豐度排名前10的物種和在屬水平上最大豐度排名前30的物種,生成物種相對豐度柱形累加圖,以便直觀查看各樣品在門和屬分類水平上相對豐度較高的物種及其比例。由圖2和圖3可知,10個樣品共獲得45個菌門,559個菌屬。
門水平上,占優勢的門主要有厚壁菌門(Firmicutes)、擬桿菌門(Bacteroidetes)、變形菌門(Proteobacteria),這3個菌門總比例在每個樣品中均在84%以上,但這3個優勢菌門在不同樣品中所占比例有所差異。厚壁菌門在10個樣品中所占比例均最高,均高于35%,其中K7樣品中高達79%。
在屬水平上,共獲得559個菌屬,其中56個屬尚未被注釋出具體的菌屬名。在10個樣品中所占比例均大于1%的菌屬有乳桿菌屬(Lactobacillus)和Romboutsia菌屬。不同菌屬在不同樣品中所占比例差異較大,其中,樣品K1和K6與其他樣品差異最為明顯。
3.4 聚類分析
為研究不同樣品間的相似性,基于Weighted Unifrac距離采用非加權組平均法(UPGMA)對樣品進行聚類分析。由圖4可知,K1和K6這2個樣品的菌群與其他樣品組成差異較大。
3 討論
傳統的分離培養方法僅限于分離能在培養基上生長的微生物,而通過該方法鑒定的微生物僅占所研究樣品中所有微生物物種的1%~10%[15]。因此,傳統的分離培養技術分析所得到的結果并不能反映所研究樣品中微生物的多樣性。近年來,Illumina MiSeq測序技術廣泛應用于各種動物腸道微生物多樣性的研究,該技術可在短時間內獲得大量數據,而且檢測數據完整性好[16]。
本研究采用Illumina MiSeq測序技術對沐川烏骨黑雞空腸微生物菌群進行分析,樣品稀釋曲線分析表明,10個樣本的覆蓋率均達到99.0%,說明此次測序數據比較全面,能夠比較全面地反映10個研究對象的空腸微生物組成情況。但10羽沐川烏骨黑雞即使在相同的飼養和管理方式下,其空腸微生物的種類和數量也不完全相同,這可能與每個檢測
個體的宿主屬性(如身體情況、飲食習慣、自身激素水平等)有關[2]。
有關腸道微生物的研究均表明,厚壁菌門(Firmicutes)、擬桿菌門(Bacteroidetes)、變形菌門(Proteobacteria)為相對豐度最高的菌門[8-9,17],本研究結果與之一致。Torok等進行了肉雞飼喂試驗,結果表明厚壁菌門、擬桿菌門、變形菌門與肉雞生產性能相關[18]。厚壁菌門在健康的火雞[19]、肉雞[9]、蛋雞[17]的腸道中也是含量最多的菌群,厚壁菌門與粗飼料的消化吸收有關,對宿主體內的物質和能量代謝有重要作用。擬桿菌菌群發酵產物有醋酸鹽,可以刺激宿主黏液相關基因的表達,促進腸道黏液的產生[20]。
在物種注釋結果中屬水平上有56個屬尚未被注釋出具體的菌屬名,這表明有關雞腸道微生物在屬水平上的研究還不夠深入。腸道微生物是一個十分復雜的系統,即使同一批在相同的飼養和管理下的雞群,不同菌屬在不同個體腸道中所占比例差異也較大,每種菌屬在宿主的新陳代謝過程中均發揮著不同作用。隨著高通量測序的廣泛應用,對腸道微生物的研究不斷深入,腸道微生物的功能研究將成為熱點,篩選出可提高宿主生產性能的有益微生物,對提高養殖經濟效益具有重要意義。
參考文獻:
[1]Gerritsen J,Smidt H,Rijkers G T,et al. Intestinal microbiota in human health and disease:the impact of probiotics[J]. Genes & Nutrition,2011,6(3):209-240.
[2]Hanning I,Diaz-Sanchez S. The functionality of the gastrointestinal microbiome in non-human animals[J]. Microbiome,2015,3(1):51.
[3]Bergmann G T. Microbial community composition along the digestive tract in forage-and grain-fed bison[J]. BMC Veterinary Research,2017,13(1):253.
[4]Barnes E M,Mead G C,Barnum D A,et al. The intestinal flora of the chicken in the period 2 to 6 weeks of age,with particular reference to the anaerobic bacteria[J]. British Poultry Science,1972,13(3):311-326.
[5]Zeng B,Han S S,Wang P,et al. The bacterial communities associated with fecal types and body weight of Rex rabbits[J]. Scientific Reports,2015,5:9342.
[6]Yu X,Wu X,Qiu L,et al. Analysis of the intestinal microbial community structure of healthy and long-living elderly residents in Gaotian Village of Liuyang City[J]. Applied Microbiology and Biotechnology,2015,99(21):9085-9095.
[7]Jia H R,Geng L L,Li Y H,et al. The effects of Bt Cry1Ie toxin on bacterial diversity in the midgut of Apis mellifera ligustica(Hymenoptera:Apidae)[J]. Scientific Reports,2016,6(1):24664.
[8]林奕岑,徐 帥,倪學勤,等. 利用Illumina MiSeq測序平臺分析肉雞盲腸微生物多樣性[J]. 中國農業大學學報,2016,21(12):65-73.
[9]徐 帥,林奕岑,周夢佳,等. 基于高通量測定肉雞回腸微生物多樣性及PICRUSt基因預測分析[J]. 動物營養學報,2016,28(8):2581-2588.
[10]Yu S,Wang G,Liao J,et al. Transcriptome profile analysis of mechanisms of black and white plumage determination in Black-Bone chicken[J]. Cellular Physiology and Biochemistry,2018,46(6):2373-2384.
[11]Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet Journal,2011,17(1):10-12.
[12]Edgar R C,Haas B J,Clemente J C,et al. UCHIME improves sensitivity and speed of chimera detection[J]. Bioinformatics,2011,27(16):2194-2200.
[13]Wang Q,Garrity G M,Tiedje J M,et al. Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy[J]. Applied and Environmental Microbiology,2007,73(16):5261-5267.
[14]Quast C,Pruesse E,Yilmaz P,et al. The SILVA ribosomal RNA gene database project:improved data processing and web-based tools[J]. Nucleic Acids Research,2013,41(1):590-596.
[15]Haraszthy V I,Zambon J J,Sreenivasan P K,et al. Identification of oral bacterial species associated with halitosis[J]. Journal of the American Dental Association,2007,138(8):1113-1120.
[16]Williams S T,Foster P G,Littlewood D. The complete mitochondrial genome of a turbinidvetigastropod from MiSeq Illumina sequencing of genomic DNA and steps towards a resolved gastropod phylogeny[J]. Gene,2014,533(1):38-47.
[17]張亞楠,魏單平,韓瑞麗,等. 高產期不同產蛋水平蛋雞腸道微生物群落特征[J]. 中國獸醫學報,2017,37(6):1179-1185.
[18]Torok V A,Hughes R J,Mikkelsen L L,et al. Identification and characterization of potential performance-related gut microbiotas in broiler chickens across various feeding trials[J]. Applied and Environmental Microbiology,2011,77(17):5868-5878.
[19]Lu J,Domingo J S. Turkey fecal microbial community structure and functional gene diversity revealed by 16S rRNA gene and metagenomicsequences[J]. Journal of Microbiology,2008,46(5):469-477.
[20]Wrzosek L,Miquel S,Noordine M L,et al. Bacteroides thetaiotaomicron and Faecalibacteriumprausnitzii influence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent[J]. BMC Biology,2013,11(1):61.陳逍遙,趙瓊瑜,彭振輝,等. 烏鱉黑色素提取工藝及其結構性狀[J]. 江蘇農業科學,2020,48(24):182-188.
