高比表面積偏高嶺土制備及其對Cr(VI)、Ni(Ⅱ)吸附性能研究
2020-05-08 05:54:46王正鄭林會牛三鑫謝曉康苗洋高峰
應(yīng)用化工 2020年3期
王正,鄭林會,牛三鑫,謝曉康,苗洋,高峰
(太原理工大學(xué) 材料科學(xué)與工程學(xué)院,山西 太原 030024)
隨著工業(yè)發(fā)展,廢水中的Cr(VI)、Ni(Ⅱ)含量超標(biāo)問題越來越顯著[1-2]。煤系高嶺土又稱高嶺石質(zhì)煤矸石,是煤系地層中的伴生礦物,作為主要的固體廢棄物之一,其資源化和高附加值引發(fā)了眾多關(guān)注[3-4]。如何實(shí)現(xiàn)“以廢治廢”,成為很多科研工作者關(guān)注的話題[5-6]。
1 實(shí)驗(yàn)部分
1.1 材料與儀器
煤系高嶺土,取自山西忻州地區(qū),具體成分見表1;K2Cr2O7、Ni(NO3)2·6H2O均為分析純。

表1 煤系高嶺土化學(xué)成分
752N紫外可見分光光度計;D2PHASER型X射線衍射儀(BRUKER);TENSOR 27紅外光譜儀(BRUKER);TM3030掃描電子顯微鏡(HITACHI);JW-BK122W比表面積及孔徑分析儀。
1.2 高比表面積偏高嶺土的制備
將煤系高嶺土樣品粉碎,60 ℃恒溫干燥12 h后放于馬弗爐中,3 h升溫到800 ℃,保溫3 h,4 h降溫到300 ℃,隨爐冷卻。
1.3 吸附實(shí)驗(yàn)
稱取1 g偏高嶺土投入100 mL吸附質(zhì)濃度為20 mg/L的錐形瓶中,置于磁力攪拌器上以(300±20)r/min攪拌,2 h后離心,吸取上清液測吸光度。其中單一吸附實(shí)驗(yàn)中吸附質(zhì)為Cr(VI)或Ni(Ⅱ),競爭吸附實(shí)驗(yàn)中吸附質(zhì)為Cr(VI)和Ni(Ⅱ)。
2 結(jié)果與討論
2.1 結(jié)構(gòu)表征
2.1.1 XRD、FTIR分析 圖1a中,高嶺土峰形尖銳對稱,晶形完整,晶體具有較高的有序度和結(jié)晶度,主要成分為高嶺石;偏高嶺土呈非晶態(tài),內(nèi)部結(jié)構(gòu)由有序完全變?yōu)闊o序狀態(tài)。
圖1b中,高嶺土在3 691,3 627 cm-1處的吸收峰由高嶺土的外羥基(表面Al—OH)、內(nèi)羥基(內(nèi)部Al—OH)自由伸縮振動形成;1 103,1 020 cm-1處峰形由Si—O垂直層、Si—O四面體片層振動形成;794,754,690 cm-1處峰形屬于Al—OH垂直振動;543 cm-1處譜帶由Al—O—Si伸縮振動形成;472 cm-1處譜帶由Si—O彎曲振動形成。
偏高嶺土在3 691,3 627 cm-1處的吸收峰消失,說明煅燒使高嶺土內(nèi)部羥基脫除;1 085 cm-1處出現(xiàn)代表Si—O伸縮振動的譜帶,823 cm-1處出現(xiàn)代表Al—O—Si振動的譜帶,466 cm-1處出現(xiàn)代表Si—O彎曲振動的譜帶。相關(guān)文獻(xiàn)[7]可知,1 085,823,466 cm-1處吸收峰為偏高嶺石特征吸收峰,結(jié)合XRD曲線可知,800 ℃煅燒后,高嶺土完全變?yōu)槠邘X的無序狀態(tài)。
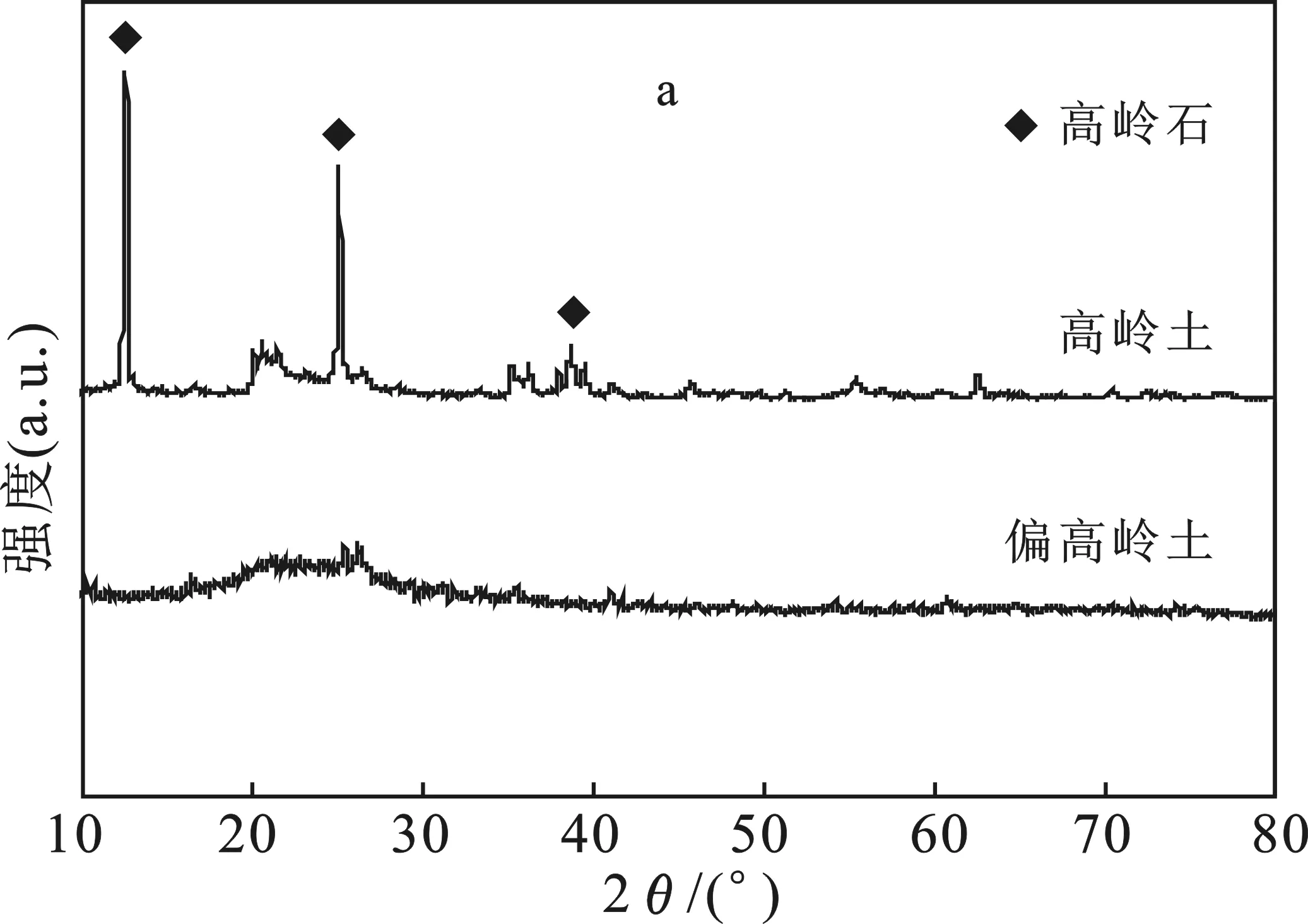
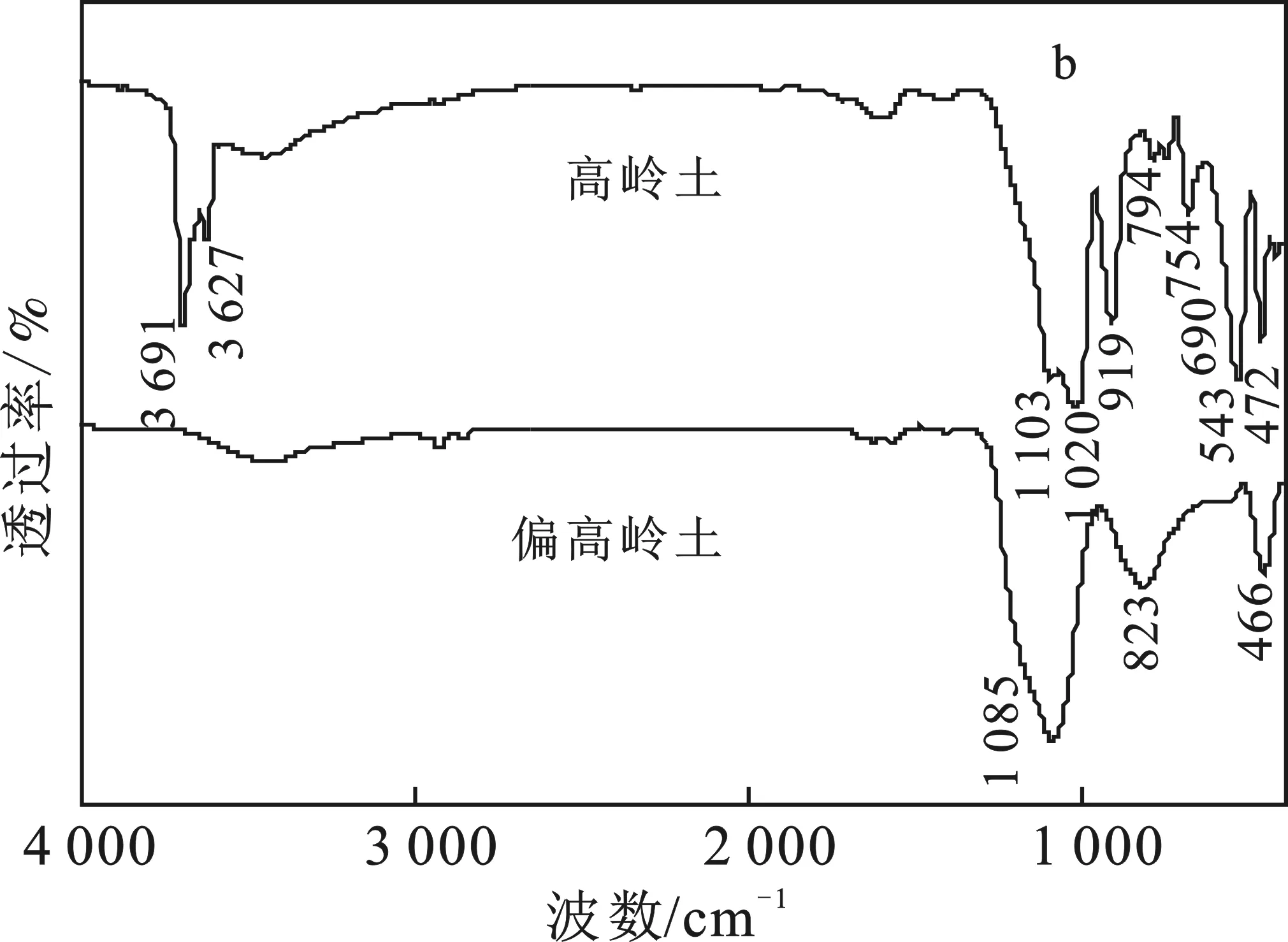
圖1 高嶺土、偏高嶺土XRD曲線(a)和FTIR曲線(b)
2.1.2 SEM分析 圖2a中,高嶺土結(jié)構(gòu)致密,排列整齊,具有明顯的片層狀結(jié)構(gòu),基本可以辨認(rèn)出正六邊形輪廓的最小單體,此時高嶺土晶型完整,結(jié)晶度高,穩(wěn)定性好;圖2b中偏高嶺土形貌雜亂無章,排列無序,片與片堆積混亂,層間孔隙增多,空隙增大,此階段的高嶺土脫羥基本完成,處于無定型狀態(tài),活性較高。

圖2 高嶺土(a)和偏高嶺土(b)的SEM圖
2.1.3 比表面積-孔結(jié)構(gòu)分析 圖3a高嶺土及圖3b 偏高嶺土均屬于IUPAC 6類吸附等溫線中可逆的Ⅱ類等溫線,即吸附類型為不受限制的單層-多層吸附。兩條曲線包含的吸附、脫附線基本重合,未存在滯后回環(huán),且低壓端部分(0.0~0.1)偏向Y軸,說明偏高嶺土與氮?dú)庵g有較強(qiáng)作用力;圖3c高嶺土及圖3d偏高嶺土在2~50 nm之間的孔徑分布表明兩者均具有介孔結(jié)構(gòu)。孔徑分布圖中兩條曲線最強(qiáng)峰對應(yīng)位置基本相同,高嶺土的最可幾孔徑為 1.07 nm,偏高嶺土的最可幾孔徑為1.06 nm。
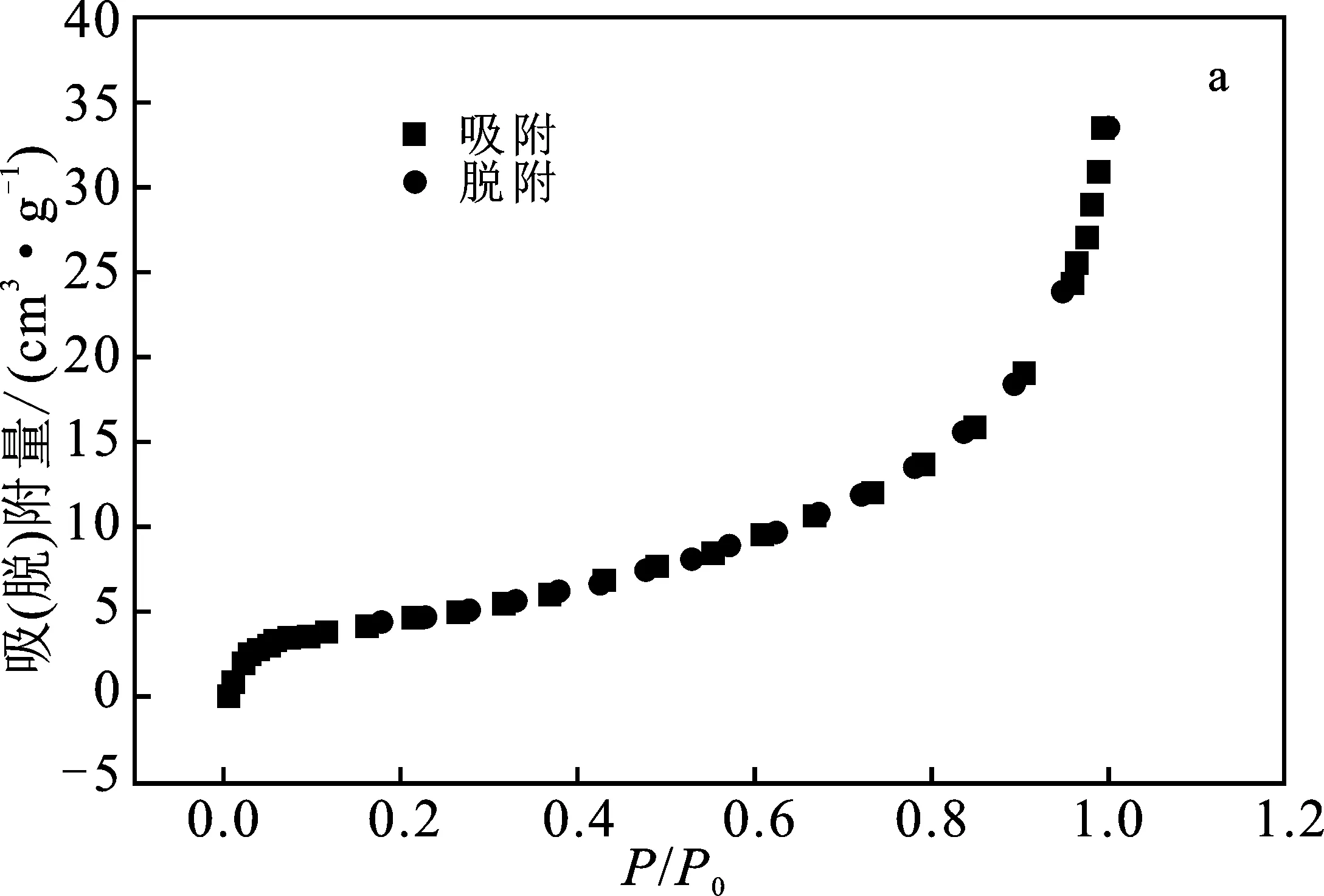

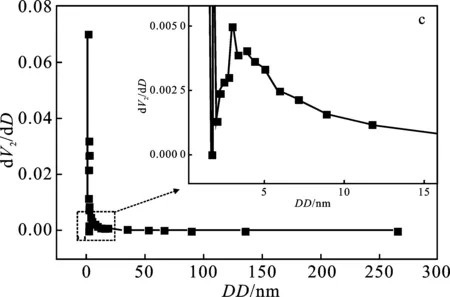
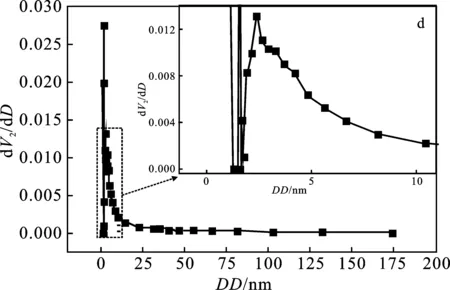
圖3 氮?dú)馕?脫附等溫線和孔徑分布曲線
表2中,偏高嶺土的比表面積、孔徑、孔體積均遠(yuǎn)大于高嶺土。綜合分析可知,煅燒后的高嶺土具有介孔結(jié)構(gòu),與氮?dú)馕侥芰υ鰪?qiáng),內(nèi)部孔徑和比表面積增加,吸附能力增大。

表2 高嶺土、偏高嶺土的BET參數(shù)
2.2 吸附時間對吸附效果的影響
圖4a,單一吸附曲線中偏高嶺土吸附Cr(VI)的反應(yīng)在120 min時趨于平衡,吸附量達(dá)到0.92 mg/g;競爭吸附曲線中該反應(yīng)在180 min時趨于平衡,吸附量達(dá)到0.91 mg/g。分析可得,Ni(Ⅱ)作為競爭離子的存在對偏高嶺土吸附Cr(VI)起到抑制作用,并且延長了吸附Cr(VI)達(dá)到的平衡時間。
圖4b中,單一吸附曲線中偏高嶺土吸附Ni(Ⅱ)的反應(yīng)在180 min時趨于平衡,吸附量達(dá)到2.69 mg/g;競爭吸附曲線中該反應(yīng)在120 min時趨于平衡,吸附量達(dá)到1.52 mg/g。分析可得,Cr(VI)作為競爭離子的存在對偏高嶺土吸附Ni(Ⅱ)起到抑制作用,但縮短了吸附Ni(Ⅱ)達(dá)到的平衡時間。
綜合分析,Cr(VI)與Ni(Ⅱ)的共存狀態(tài)會抑制對其中單一離子的吸附,偏高嶺土對Ni(Ⅱ)的吸附量大于對Cr(VI)的吸附量。
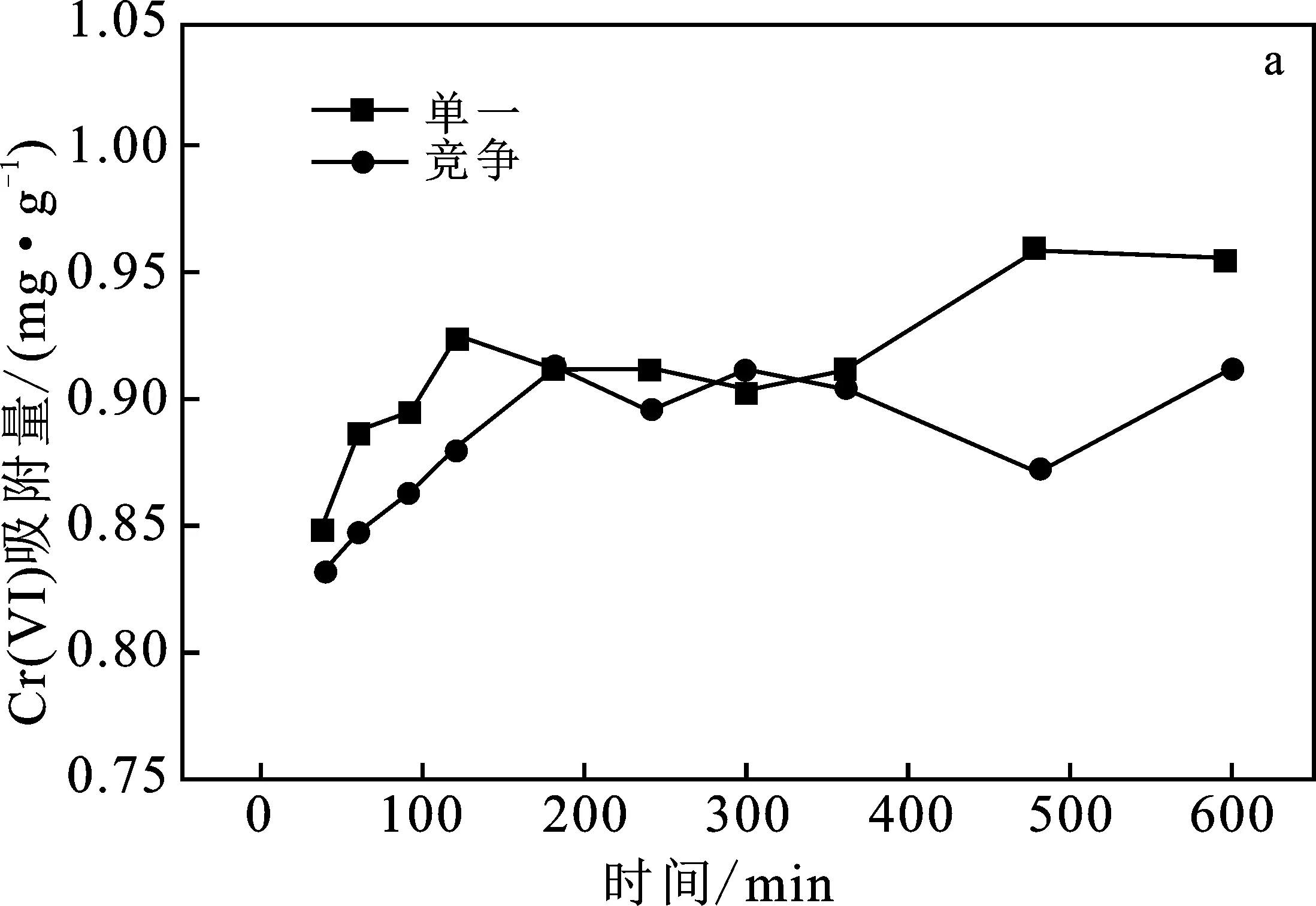
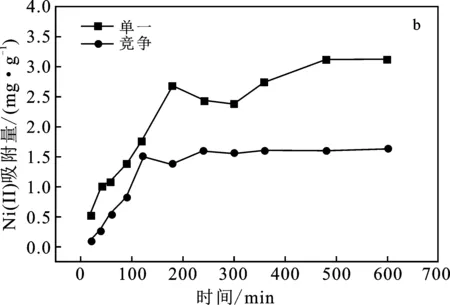
圖4 吸附時間對Cr(VI)、Ni(Ⅱ)吸附量的影響
2.3 pH值對吸附效果的影響
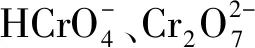
圖5b單一吸附和競爭吸附中,偏高嶺土對Ni(Ⅱ)的吸附量隨pH值升高呈上升趨勢,當(dāng)pH值>8時吸附量接近最大。分析可得,pH值<7時,體系中H+較多,與Ni(Ⅱ)形成競爭,導(dǎo)致偏高嶺土對Ni(Ⅱ)的吸附量減少;pH值>7時,體系中H+減少,致使對Ni(Ⅱ)的吸附量逐漸增加。所以pH值>6,偏高嶺土對Ni(Ⅱ)的吸附性能更好。
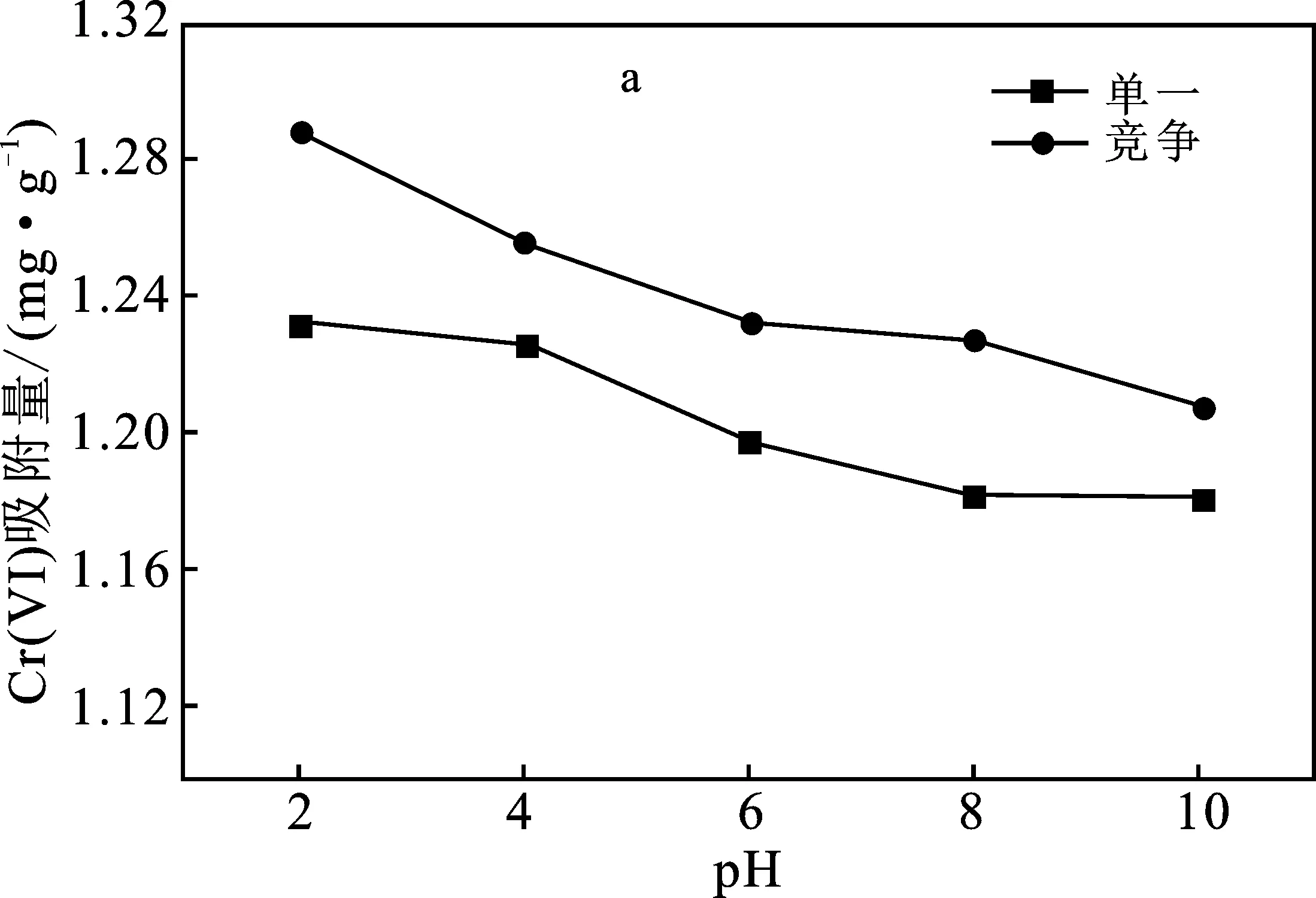
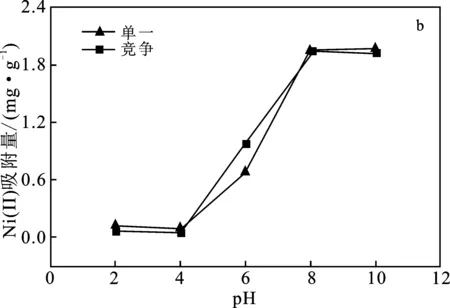
圖5 pH值對Cr(VI)、Ni(Ⅱ)吸附量的影響
2.4 NaCl濃度對吸附效果的影響
圖6a單一和競爭條件下,隨著NaCl溶液濃度的升高,偏高嶺土對Cr(VI)的吸附量總體呈現(xiàn)上升;圖6b單一和競爭條件下,隨著NaCl溶液濃度的提高,偏高嶺土吸附Ni(Ⅱ)的量不斷下降。綜合分析,NaCl溶液的加入促進(jìn)了偏高嶺土對Cr(VI)的吸附,極大地抑制了偏高嶺土對Ni(Ⅱ)的吸附。


圖6 NaCl濃度對Cr(VI)、Ni(Ⅱ)吸附量的影響
2.5 物理化學(xué)特性對吸附效果的影響
表3中,Ni離子半徑比Cr大,對外層電子的束縛能力相對較弱,而水和離子半徑較小,所以容易與偏高嶺土結(jié)合。Ni電負(fù)性較大,吸引共有體系中電子的能力更強(qiáng),且荷徑比較小,更容易與偏高嶺土內(nèi)部活性位點(diǎn)形成共價鍵或絡(luò)合物。分析可知,Ni的物化特性優(yōu)勢導(dǎo)致偏高嶺土對其的吸附量更高。

表3 鉻元素、鎳元素的物理化學(xué)特性參數(shù)
3 結(jié)論
(1)通過預(yù)定煅燒可制備出高比表面積偏高嶺土。微觀分析可得,處理后的偏高嶺土具有介孔結(jié)構(gòu),對氮?dú)獾奈?脫附為不受限制的單層-多層吸附。偏高嶺化過程中,由于脫羥反應(yīng)的發(fā)生,高嶺土比表面積增大,總孔體積增多,片層狀結(jié)構(gòu)破壞,內(nèi)部由結(jié)晶度很高的有序狀態(tài)變?yōu)榉蔷ЫY(jié)構(gòu)的無序狀態(tài),活性增加,可作為良好的吸附劑使用。
(2)偏高嶺土對Cr(VI)的吸附實(shí)驗(yàn)中,pH值<6 時吸附性能更好,在體系中加入NaCl溶液能促進(jìn)對Cr(VI)的吸附;偏高嶺土對Ni(Ⅱ)的吸附實(shí)驗(yàn)中,pH值>6時吸附效果更好,在體系中加入NaCl溶液極大阻礙了偏高嶺土對Ni(Ⅱ)的吸附。
(3)偏高嶺土對Cr(VI)、Ni(Ⅱ)共存體系的吸附過程中,競爭狀態(tài)的存在抑制了對其中單一離子的吸附。
(4)Cr、Ni的物化特性表明,Ni的離子半徑較大,對外層電子的束縛能力相對較弱,其水合離子半徑較小,電負(fù)性較大,荷徑比較小,在吸附過程中更容易在偏高嶺土內(nèi)部形成共價鍵或絡(luò)合物,從而比Cr吸附效果更好。
猜你喜歡
科學(xué)大眾(2023年17期)2023-10-26 07:39:14
哲學(xué)評論(2021年2期)2021-08-22 01:53:34
民用飛機(jī)設(shè)計與研究(2020年4期)2021-01-21 09:15:02
天天愛科學(xué)(2020年6期)2020-09-10 07:22:44
中華詩詞(2019年7期)2019-11-25 01:43:04
電子制作(2018年18期)2018-11-14 01:48:24
數(shù)學(xué)物理學(xué)報(2017年6期)2018-01-22 02:26:40
山東工業(yè)技術(shù)(2016年15期)2016-12-01 05:31:22
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
現(xiàn)代企業(yè)(2015年9期)2015-02-28 18:56:50
