熒光衍生化-磁分散固相萃取/高效液相色譜檢測池塘水與蔬菜中3種揮發性異味物質
2020-05-08 13:40:42鄭振佳董京磊孫魯平朱樹蕓趙先恩
分析測試學報 2020年3期
陳 坤,張 鵬,王 妍,鄭振佳,董京磊,孫魯平, 朱樹蕓,趙先恩*
(1.曲阜師范大學 化學與化工學院,山東省綠色天然產物與醫藥中間體高校重點實驗室,山東 曲阜 273165; 2.山東農業大學 食品科學與工程學院,山東 泰安 271018;3.龍大食品集團有限公司,山東 煙臺 265200)
水體中的醇類揮發性異味物質(VOCs)主要包括2-甲基異莰醇(2-MIB)、土臭素(GSM)以及3-甲基-1-丁醇(3-MB)。隨著人們對VOCs研究的不斷深入,發現VOCs是產生水體污染的原因之一,且會影響飲用水、食品、養殖魚類的味道和口感。VOCs在各種水體中含量極低,通常無法通過嗅覺判斷其含量。因此,建立一種對VOCs進行高靈敏檢測及質量控制的方法具有重要意義[1-3]。
已報道的VOCs等揮發性有機物的檢測方法主要有氣相色譜法(GC)、氣相色譜-質譜法(GC-MS)以及分子印跡熒光位移法[4-8]。其中,GC法通常采用火焰光度檢測器、電子捕獲檢測器和火焰離子檢測器(GC-FID)對VOCs進行檢測,但靈敏度較低且基質效應嚴重。GC-MS法報道較多,但檢測靈敏度較低且儀器昂貴。分子印跡熒光位移法對VOCs的檢測雖具有特異性,但靈敏度較低且制備過程繁瑣。此外,VOCs的樣品前處理方法較多,包括閉環捕集(CLSA)、液液微萃取(LLME)、吹掃捕集(P&T)、固相微萃取(SPME)等。但上述樣品前處理技術對VOCs的研究結果往往難以令人滿意,或操作較為復雜,或使用成本較高。目前雖有分析2-MIB和GSM的少量報道,但鮮有同時分析2-MIB、GSM與3-MB的研究。
對于食品和環境分析,衍生化結合前處理技術能簡化樣品的制備、降低基質效應、提高檢測靈敏度,對食品安全監控和水體污染物的監測有重要意義[9]。磁分散固相萃取(MDSPE)技術可以選擇性吸附小分子有機物的衍生化產物,且萃取后易于分離收集,操作快速簡便[10]。氧化石墨烯具有超大的比表面積和良好的表面修飾性,可與化合物形成氫鍵、π-π堆積等作用[11]。使用磁性氧化石墨烯(MGO)進行MDSPE同時具有強吸附性和磁分離能力。因此,本研究建立了以6-碳酰氯左氧氟沙星(LFC-Cl)為衍生化試劑,MGO為MDSPE吸附劑的高效液相色譜-熒光檢測(HPLC-FLD)分析方法。本方法實現了池塘水和蔬菜中前述3種VOCs的檢測,具有靈敏度高、樣品前處理簡單、儀器普適性好等優勢。
1 實驗部分
1.1 儀器與試劑
Agilent 1260高效液相色譜儀(配備四元梯度泵、在線真空脫氣機、熒光檢測器、自動進樣器、恒溫柱溫箱,美國Agilent公司),RE-2000B旋轉蒸發儀(上海亞榮生化儀器廠),KQ2200E超聲波清洗器(江蘇昆山超聲儀器有限公司),DF-2集熱式磁力攪拌器(金壇市成輝儀器廠),真空干燥箱、高速萬能粉碎機(北京市永光明醫療儀器有限公司),SC-06低速離心機(安徽中科中佳科學儀器有限公司),XW-80A旋渦混勻器(上海精科實業有限公司),SHA-C水浴恒溫振蕩器(金壇市金南儀器制造有限公司)。
左氧氟沙星(LFC,98.0%)、甲酸(色譜級,98.0%)、無水級1,2-二氯乙烷(99.8%)、4-二甲氨基吡啶(DMAP,99%)均購自阿拉丁公司,乙腈(99.9%,北京百靈威科技有限公司),三氯氧磷(POCl3,山東西亞化學股份有限公司),2-MIB(100 mg/L,德國Dr.Ehrenstorfer GmbH),GSM(10 ng/L,英國LGC集團),3-MB(99.0%,梯希愛化成工業發展有限公司),其他試劑均為分析純。MGO參照本實驗室方法合成[10]。
1.2 LFC-Cl的合成
LFC-Cl的合成參照文獻[12-13]部分合成條件,并進行合成方案修改:向三頸燒瓶中加入1.0 g LFC和60 mL無水級1,2-二氯乙烷,用移液槍緩慢滴加2.2 mL POCl3,于85 ℃回流反應4 h。冷卻后加入0.05 g活性炭,于85 ℃再次回流20 min。趁熱過濾,除去活性炭,旋蒸除去溶劑得到LFC-Cl,置于真空烘箱中干燥8 h。干燥后的LFC-Cl產物可直接用于衍生化反應。
1.3 溶液配制
量取適宜體積的2-MIB、GSM、3-MB標準品,分別溶于乙腈得濃度為1.91×10-5mol/L的標準溶液,相應的低濃度溶液均由此標準溶液用乙腈稀釋得到。取一定體積的3種標準溶液配成濃度為4.78×10-6mol/L的混合標準溶液。準確稱取一定量DMAP溶于乙腈,得到濃度為0.20 mol/L的DMAP溶液。
1.4 衍生化反應
準確稱取6 mg LFC-Cl于安瓿瓶中,加入250 μL乙腈、400 μL待測物混合標準溶液(或實際提取樣品)、350 μL 0.05 mol/L的DMAP標準溶液,封口后,在60 ℃水浴中超聲波輔助條件(超聲波功率100 W,頻率40 kHz)下衍生反應90 min。衍生化反應示意圖見圖1。衍生溶液待進行MDSPE過程。
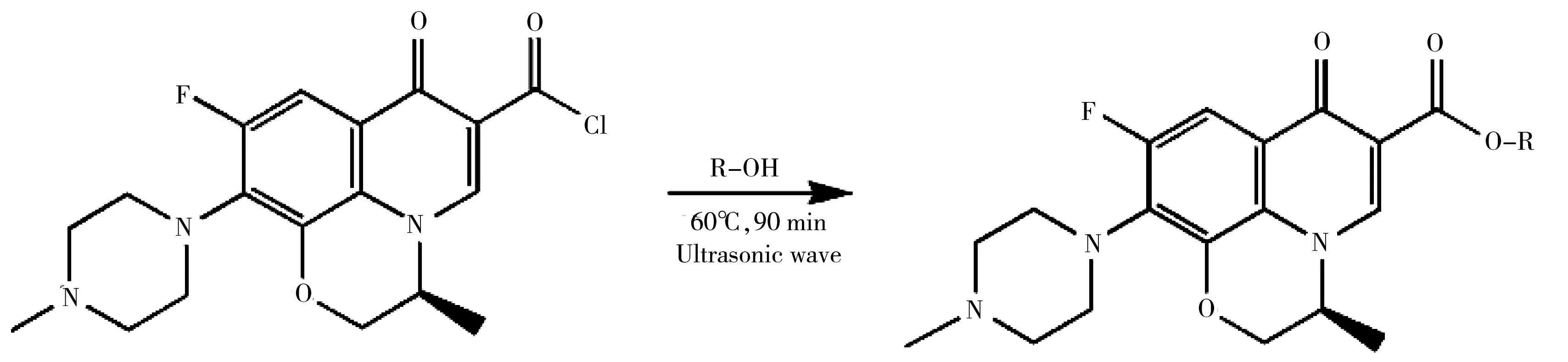
圖1 6-碳酰氯左氧氟沙星與含羥基揮發性異味物質的衍生化過程Fig.1 Derivatization reaction between LFC-Cl and hydroxyl-containing volatile organic compounds
1.5 MDSPE條件
MDSPE條件參考文獻的部分實驗方法[10,14],并修改如下:將20 mg MGO加入衍生溶液后,置于25 ℃水浴恒溫振蕩器中振蕩20 min,在磁鐵作用下分離棄去上清液,用1 mL乙腈洗滌3次,加入1 mL洗脫劑乙腈(含1%甲酸),在超聲波輔助條件(超聲波功率100 W,頻率40 kHz)下解吸3 min,在磁鐵作用下分離上清液,定容至1 mL,過0.45 μm有機相濾膜后稀釋進樣20 μL進行HPLC-FLD分析。
1.6 HPLC條件
色譜柱(CC):Agilent ZORBAX SB-C18柱(4.6 mm×150 mm,5 μm,美國Agilent公司)。流動相:A相為10%乙腈水溶液(含0.1%甲酸),B相為乙腈(含0.1%甲酸)。梯度洗脫程序(GE):0~6 min,2%B;6~10 min,2%~100%B;10~15 min,100%B。流速:1.0 mL/min,進樣量:20 μL,柱溫:30 ℃。熒光激發波長(λex)和發射波長(λem)分別為295、460 nm。
1.7 實際樣品處理
實際樣品處理參照文獻的部分處理方法[7,12,15-16]。
1.7.1 池塘水處理量取50 mL池塘水于分液漏斗中,加入5.0 g 氯化鈉固體,加入300 μL氯仿作萃取劑,萃取3次合并萃取液。用無水硫酸鈉脫水干燥后定容至1 mL,待衍生。
1.7.2 蔬菜樣品處理準確稱取15.0 g綠色蔬菜(生菜、萵苣、蔥)置于萬能粉碎機打碎,收集后加入7.5 mL乙腈渦旋2 min,超聲提取20 min。加入1.0 g氯化鈉和4.0 g無水硫酸鎂固體,渦旋5 min,以5 000 r/min 脫水15 min,待分層后準確吸取乙腈層,再次脫水后吸取乙腈層,待衍生化反應。
2 結果與討論
2.1 HPLC條件的優化
實驗對比了不同色譜柱(CC1:Agilent ZORBAX 300SB-C18柱(4.6 mm×250 mm,5 μm);CC2:Agilent ZORBAX SB-C18柱(4.6 mm×150 mm,5 μm);CC3:依利特 SinoChrom ODS-BP(4.6 mm×250 mm,5 μm);CC4:依利特HYPERSL C18(4.6 mm×100 mm,5 μm)),以及調整不同GE的分離效果,相同條件下平行測定3次(見圖2)。對CC考察結果表明,采用CC2分離時3種VOCs衍生物的峰面積最大(圖2A)。對GE考察結果表明,采用GE4時3種VOCs衍生物的峰面積最大(圖2B),由于B相(含0.1%甲酸的乙腈)在0~6 min內已為2%,且能夠實現3種VOCs衍生物的基線分離,因此采用GE4作為最佳梯度洗脫程序。采用Agilent 1260 HPLC-FLD儀器的在線熒光光譜掃描功能,確定3種VOCs衍生物HPLC-FLD的最佳激發和發射波長分別為295 nm和460 nm。最終確定了“1.6”所述色譜條件,3種VOCs衍生物在15 min內可實現基線分離(見圖3)。
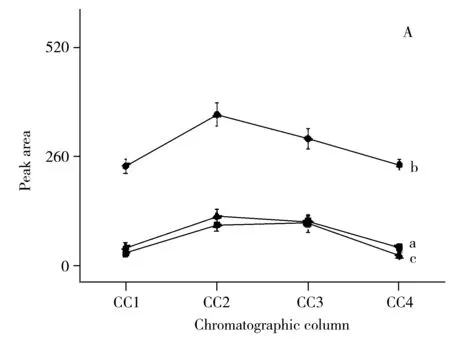
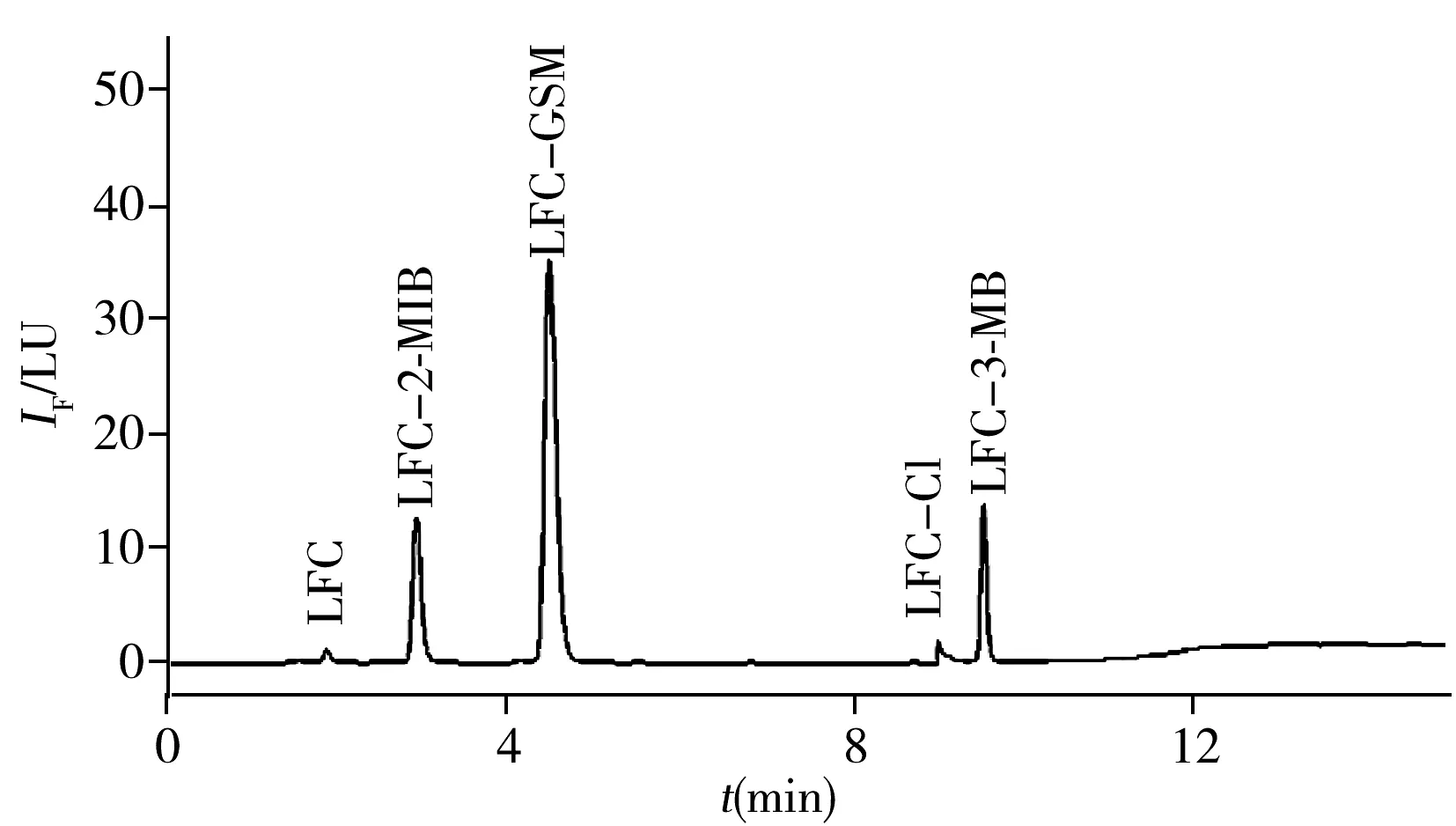
圖3 3種揮發性異味物質衍生物的高效液相色譜圖Fig.3 HPLC chromatogram of three derivatives of volatile organic compounds LFC:levofloxacin(左氧氟沙星);LFC-2-MIB:derivatives of 2-methylisoborneol(左氧氟沙星-2-甲基異莰醇衍生物);LFC-GSM: derivatives of geosmin(左氧氟沙星-土臭素衍生物);LFC-Cl: 6-carbonyl chloride levofloxacin(6-碳酰氯左氧氟沙星); LFC-3-MB:derivatives of 3-methyl-1-butanol(左氧氟沙星-3-甲基-1-丁醇衍生物)
2.2 衍生化條件的優化
實驗同時考察了衍生化反應溫度、反應時間、DMAP濃度以及衍生試劑用量對3種衍生物峰面積的影響,得到揮發性異味物質衍生化溶液后分別平行測定3次。
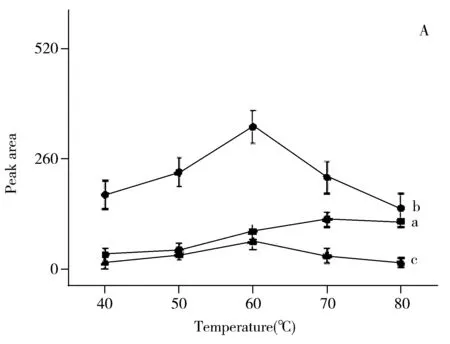
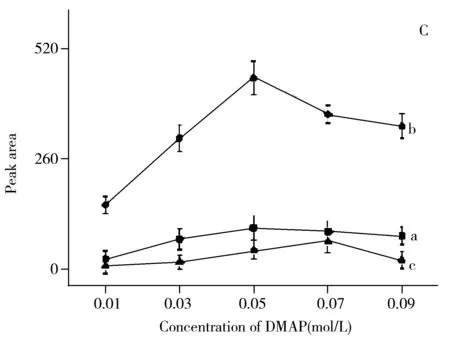
對反應溫度和反應時間的考察結果表明:衍生物峰面積隨著反應溫度升高和反應時間的延長而增加,在60 ℃、90 min時峰面積達最大;繼續升高反應溫度或延長反應時間,衍生物的峰面積均減小(圖4A和4B)。這可能是由于反應溫度過高、反應時間過長使衍生產物發生分解所致。對DMAP濃度的考察結果表明:當DMAP濃度低于0.05 mol/L時,衍生物峰面積隨其濃度的增大而增加;DMAP濃度為0.05 mol/L時衍生物的峰面積達最大;但當DMAP濃度大于0.05 mol/L后,衍生物的峰面積隨其濃度的增加而減小(圖4C)。這可能是由于催化劑DMAP的濃度增大導致反應產物在強堿性條件下分解。對衍生試劑用量的考察結果表明:衍生物的峰面積隨衍生試劑用量的增加而增大,當衍生試劑用量為3種VOCs用量的1 500倍時,衍生物的峰面積最大,但繼續增大衍生試劑用量,峰面積反而減小(圖4D)。最終確定衍生化條件如“1.4”所述。
2.3 MDSPE條件的優化
實驗同時考察了MGO吸附劑用量(10、15、20、25、30 mg)、萃取時間(10、15、20、25、30 min)、解吸時間(1、2、3、4、5 min)和解吸劑種類(均含1%甲酸的甲醇、乙醇、乙腈、丙酮)對萃取率的影響,得到異味物質衍生化溶液后分別平行測定3次。對吸附劑用量的考察結果表明:萃取率隨著吸附劑用量的增加而增加,當吸附劑用量為20 mg時,萃取率最大;繼續增加吸附劑用量,萃取率并無明顯增大。對萃取時間的考察結果表明:萃取率隨萃取時間的延長而增加,萃取時間為20 min時萃取率最大;繼續延長反應時間,萃取率并無明顯增大。對解吸時間的考察結果表明:萃取率隨解吸時間的延長而增大,解吸時間為3 min時萃取率最大;繼續延長解吸時間,萃取率并無明顯增大。對解吸劑種類的考察結果表明:以乙腈(含1%甲酸)作解吸劑時的萃取率最大,這可能是不同解吸劑的極性、目標分子溶解度等存在較大差異,乙腈(含1%甲酸)能夠更好地將異味物質衍生物從MGO中洗脫出來。最終確定MDSPE優化條件如“1.5”所述。
研究表明[11],MGO對有機化合物的吸附機理主要是吸附劑與被吸附分子形成氫鍵、π-π堆積、靜電作用力、范德華力以及疏水作用等。本方法所使用的衍生試劑分子中存在可形成氫鍵的羰基、醚鍵、氟原子和氮原子等以及較大的共軛體系等結構,可與吸附劑形成上述作用力,因而產生良好的萃取效果。
2.4 方法評價
2.4.1 線性、檢出限、定量下限與精密度在空白水樣氯仿溶液中添加標準樣品,按照衍生化方法和MDSPE過程進行處理,根據峰面積(Y)和實際進樣濃度(X)進行線性回歸,并分別以信噪比S/N>3和S/N>10得到3種衍生物的檢出限(LOD)和定量下限(LOQ)。為便于與其他方法及文獻中VOCs的檢測結果進行對比,將3種VOCs衍生物的數據換算為VOCs的數據,結果見表1。結果表明,3種VOCs在一定質量濃度范圍內線性關系良好,相關系數r≥0.987,LOD為0.020~0.95 ng/L,LOQ為0.10~3.3 ng/L。已有文獻報道2-MIB和GSM的嗅味閾值約為10 ng/L[3],本方法測得3種VOCs的LOD和LOQ均低于VOCs的嗅味閾值,表明本方法可用于極低含量的VOCs樣品檢測。
在相同實驗條件下,制備混合標準品衍生溶液,平行6次進樣分析,得到3種VOCs衍生物保留時間和峰面積的相對標準偏差(RSD)均不大于1.7%(表1)。結果表明,本方法的精密度良好。
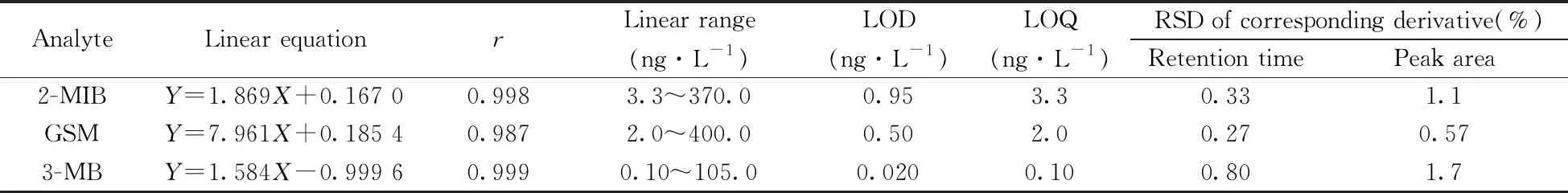
表1 3種異味物質的線性方程、相關系數、線性范圍、檢出限、定量下限與相對標準偏差(n=6)Table 1 Linear equations,correlation coefficients(r),linear ranges,LODs,LOQs and RSDs of three VOCs(n=6)
Y:peak area;X:injection concentration(ng/L)
2.4.2 與文獻方法的比較與文獻方法相比,本方法所用的衍生試劑LFC-Cl易于合成并首次被應用于VOCs檢測。在檢出限、操作難易程度和儀器普適性等方面的對比見表2。
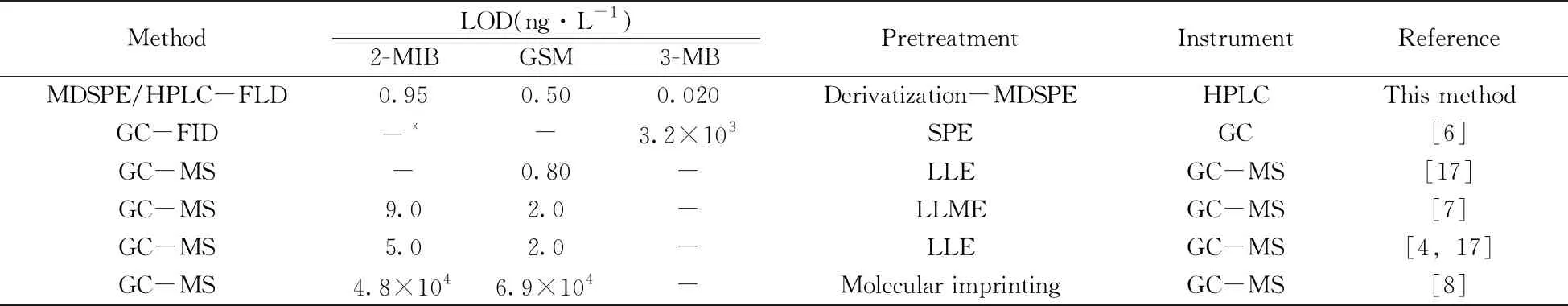
表2 本方法與文獻方法的比較Table 2 Comparisons of this method with the reported methods
*not reported(文獻中未對此種物質進行檢測)
結果顯示,在檢出限方面,本方法的檢出限低于或與LLE/GC-MS[4,17]和LLME/GC-MS[7]法的檢出限相近;而遠低于SPE/GC法[6]和分子印跡熒光位移/GC-MS法[8]。這一方面是由于本方法使用的衍生試劑LFC-Cl具有較大的共軛體系,從而為高靈敏檢測VOCs帶來良好的效果。另一方面,MDSPE技術能夠選擇性地吸附小分子有機物的衍生化產物,起到富集和凈化作用。在操作難易程度上,本方法使用的MDSPE與LLME、SPE等前處理方法相比無需特定設備,成本較低,且操作較簡單;MGO的制備步驟比分子印跡材料少,且適用于多種異味物質衍生物的同時吸附。在儀器普適性方面,HPLC-FLD的普適性較好。
2.5 回收率與實際樣品應用
以池塘水(取自曲阜市某池塘)和蔬菜(生菜、萵苣、蔥,購自曲阜某菜市場)為實際樣品,采用本方法進行前處理和實驗,同時進行加標回收率實驗。由表3可知,3種VOCs的回收率為76.8%~118%,能滿足實際檢測要求。實驗發現,蔬菜樣品中VOCs的含量較低,而池塘水中VOCs的含量較高。池塘水和蔬菜樣品中3種VOCs衍生物的色譜圖見圖5。另外開展了對自來水、紫菜、海帶等的適用性考察,測定結果的RSD均小于12%(n=5)。表明本方法的適用性良好,能夠應用于各種實際樣品中異味物質的檢測。
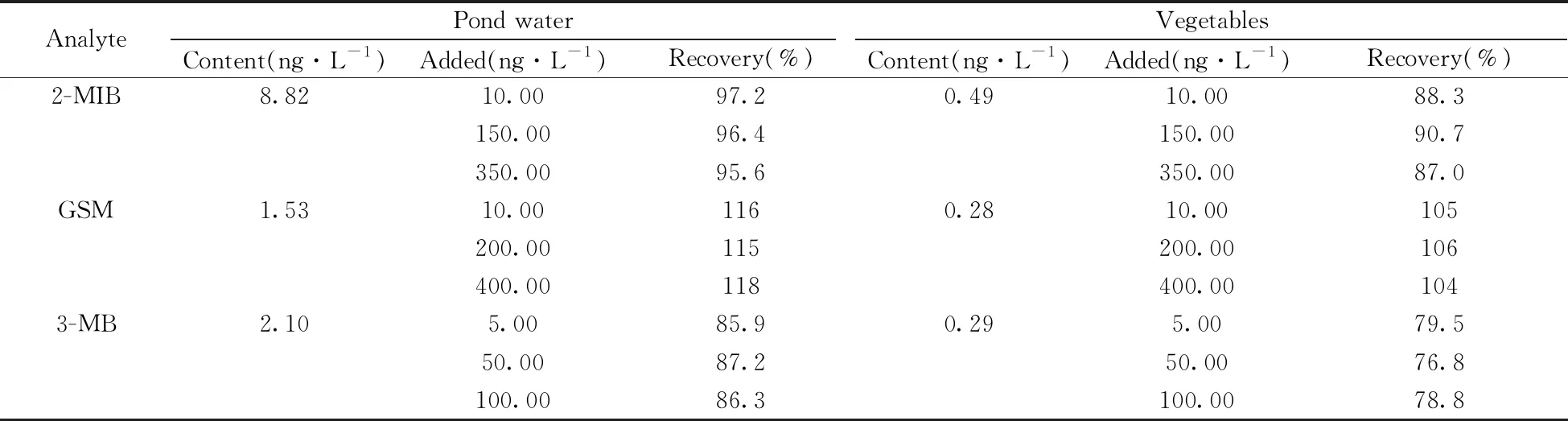
表3 兩種實際樣品中異味物質的含量及加標回收率(n=3)Table 3 Contents and recoveries of VOCs in two practical samples(n=3)
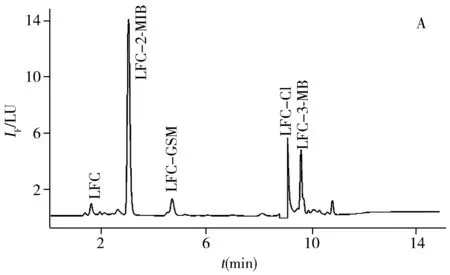
圖5 池塘水樣(A)與蔬菜樣品(B)中3種異味物質衍生物的色譜圖
Fig.5 Chromatograms of the three derivatives of VOCs in pond water(A) and vegetables(B)
3 結 論
本文利用衍生化技術結合磁固相萃取技術,建立了3種VOCs的HPLC-FLD分析方法。該方法具有靈敏度高、樣品前處理簡單、儀器普適性好等優勢。實際檢測結果表明,蔬菜中含有較少的VOCs,而池塘水中VOCs的含量相對較多。因此,需加強對各種湖泊池塘的管理和凈化,以降低VOCs對人類健康的危害,減少對生態環境的破壞。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
