苯并芘DNA加合物anti-BPDE-N2-dG四種立體異構體的合成及色譜分離
2020-05-08 13:40:42趙旭霞郭婭男鄭速進張加玲閻小青
分析測試學報 2020年3期
關鍵詞:檢測
趙旭霞,馮 蕊,郭婭男,鄭速進,張加玲,閻小青
(山西醫科大學 公共衛生學院 衛生檢驗教研室,山西 太原 030001)
多環芳烴(PAHs)是一類廣泛存在于環境的持久性有機污染物,主要來源于煤、石油等有機化合物的不完全燃燒,也會產生于燒烤食物以及瀝青、焦油等的生產過程中。其中,苯并[a]芘(B[a]P)是最為典型的一種多環芳烴,具有極強的致畸、致癌和致突變性,在進入人體后經過一系列代謝活化作用生成鄰二醇環氧苯并芘(BPDE)[1-3],此代謝產物有(±)anti-BPDE和(±)syn-BPDE兩對對映異構體(anti-BPDE與syn-BPDE之間為非對映異構體)。大量的研究表明(±)anti-BPDE比其立體異構體(±)syn-BPDE更具誘變性[4-7],(±)anti-BPDE主要與生物體DNA中脫氧鳥苷的N-2位點結合[1-4],生成BPDE的脫氧鳥苷加合物(anti-BPDE-N2-dG)。此加合物是化學物質致突變、致癌進程中一個重要的啟動因子[8],既可以作為暴露標志物,反映毒物在靶位的實際暴露劑量,又可以作為效應標志物,反映DNA受到有毒化學物質損傷的程度,對研究環境和職業B[a]P暴露的早發現、早預防有重要意義[9-10]。anti-BPDE-N2-dG含有兩個手性碳原子,存在對映異構和順反異構現象,主要有trans(-)、trans(+)、cis(+)、cis(-)-anti-BPDE-N2-dG四種立體異構(如圖1所示)[11],其中,trans(-)與trans(+)-anti-BPDE-N2-dG,cis(+)與cis(-)-anti-BPDE-N2-dG為兩對對映異構體(手性異構體),而trans與cis-anti-BPDE-N2-dG為順反異構體。研究表明這四種立體異構體的生物學活性存在顯著差異[12]。因此,對anti-BPDE-N2-dG加合物的四種立體異構體進行準確地定性和定量分析,對B[a]P暴露監測及其致癌、致突變的作用機理研究非常重要。
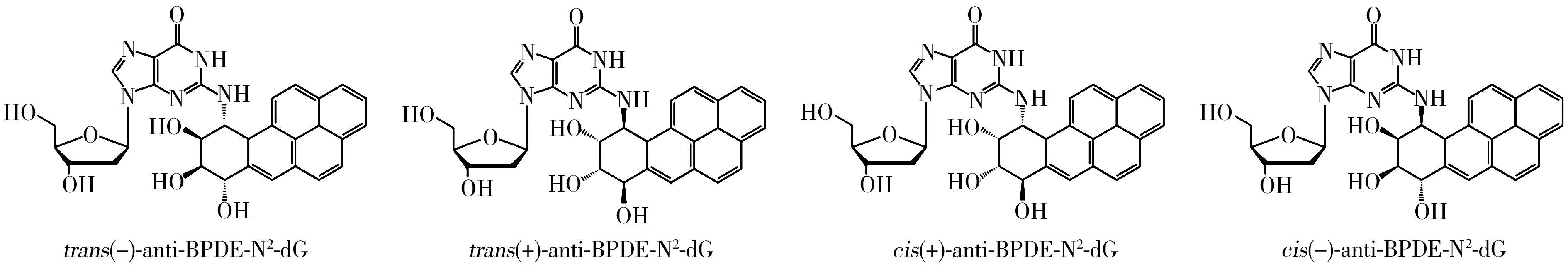
圖1 anti-BPDE-N2-dG立體異構體的分子結構Fig.1 Molecular structures of anti-BPDE-N2-dG stereoisomers
環境中多環芳烴的暴露水平極低,所以暴露后的生物體內相應形成的DNA加合物含量更低。大約108~1010個核苷酸中含有1個加合物[4],如何從大量的正常核苷中檢出痕量的特定加合物是當今分析技術面臨的挑戰。目前,關于苯并芘DNA加合物的檢測發展了多種方法[13-14],包括32P標記法、免疫吸附法、核磁共振法、熒光檢測法,但這些方法均無法對加合物的立體異構體進行表征。相比之下,高效液相色譜-質譜聯用法(HPLC-MS/MS)不僅擁有可與32P后標記法相媲美的靈敏度,而且還可以提供加合物的結構信息,因此受到了更多關注[15-19],但在使用該方法定量檢測BPDE-DNA加合物時,需合成相應的標準品。目前,文獻報道了BPDE-DNA加合物的不同合成方法[15-18],其中,王海林課題組[15]創新性地通過anti-BPDE與2′-脫氧鳥苷(Deoxyguanosine,dG)直接反應后,用固相萃取(SPE)和高效液相色譜(HPLC)對反應產物進行提純制得anti-BPDE-N2-dG四種立體異構體。該方法與以往的合成方法相比,具有無需酶解、反應時間短、BPDE用量少等優點。但該合成方法利用常規苯基柱需要85~100 min[15]才可將anti-BPDE-N2-dG四種立體異構體實現色譜分離提純。另外,在利用HPLC-MS/MS對anti-BPDE-N2-dG四種立體異構體進行檢測時,雖然質譜的選擇性監測可排除副產物四醇-B[a]P的干擾,縮短色譜分離時間,但仍需45~60 min才能將四種立體異構體分離并檢測[18-19],且流速快,分流檢測使得樣品用量大,分離度較小。
五氟苯基柱(PFP)是在硅膠基質上鍵合的五氟苯基硅烷鍵合相,由于五氟苯基的存在,化合物可與鍵合相之間產生π-π共軛、偶極-偶極、氫鍵等作用[20],因此PFP柱的選擇性有別于C18柱和苯基柱。本研究首次將PFP應用于anti-BPDE-N2-dG四種立體異構體的分離,結果表明,相對于常規C18色譜柱,anti-BPDE-N2-dG 四種立體異構體在 PFP色譜柱上的分離效果更好。通過對色譜條件的優化,可實現45 min內將四種立體異構體分離提純。在使用HPLC-MS/MS檢測時,30 min內即可完成檢測,極大提高了分離與檢測效率。
1 實驗部分
1.1 儀器與試劑
高效液相色譜配二極管陣列(DAD)檢測器(Thermo scientific U3000,美國Thermo公司);高效液相色譜-串聯質譜(LCMS-8050,日本島津公司);MOS 450圓二色光譜儀(法國BioLogic公司);TU-1901雙光束紫外分光光度計(北京普析通用儀器有限公司);SCIENTZ-12N冷凍干燥機(寧波新芝生物科技股份有限公司);GI36DS高壓滅菌鍋(廈門致微儀器有限公司);Mikro 220R高速冷凍離心機(德國Hettich公司);PB-10 pH計(德國Sartorius公司);ME104E電子天平(瑞士Mettler-Toledo公司);RCT basic加熱磁力攪拌器(德國IKA集團)。
(±)anti-BPDE(純度>99%,美國MRIGlobal Chemical Carcinogen Repository);2′脫氧鳥苷(純度98%,百靈威科技有限公司);小牛胸腺DNA(美國Sigma公司);脫氧核糖核酸酶Deoxyribonuclease I from bovine pancreas(美國Sigma公司);蛇毒磷酸二酯酶Phosphodiesterase I from Crotalus adamanteus venom(美國Sigma公司);Tris-base(上海Sangon Biotech);四氫呋喃、三乙胺、氯化鈉、氯化鈣、乙醚、無水乙醇、乙酸乙酯、濃鹽酸(分析純,天津市光復精細化工研究所);六水氯化鎂(上海BBI Life Sciences);甲醇、乙腈(色譜純,美國Thermo Fisher公司);甲酸(色譜純,天津市致遠化學試劑有限公司);超純水(屈臣氏飲用水)。
1.2 anti-BPDE-N2-dG四種立體異構體標準品的制備
1.2.1 合成反應及固相萃取的初步純化(±)anti-BPDE與小牛胸腺DNA反應生成BPDE-脫氧鳥苷酸化合物(BPDE adduct of deoxyguanosine monophosphate,BPDE-dGMP):參照Vouros[21]課題組的方法合成BPDE-dGMP。稱取 0.9 mg(±)anti-BPDE溶于1 mL四氫呋喃;稱取3.6 mg小牛胸腺DNA溶于5 mL 0.05 mol/L Tris-HCl(pH 6.8);將兩者混勻,50 ℃避光孵育8 h。孵育結束,加乙酸乙酯-乙醚(5∶2,體積比)7 mL液液萃取,收集下層液體,重復3次。加入NaCl使其在溶液中的濃度為0.14 mol/L,搖勻,再加入2.5倍體積的無水乙醇,充分混勻,-22 ℃靜置20~30 min后,-10 ℃ 12 000 r/min離心20 min,移走上清液,室溫下干燥,復溶于0.05 mol/L含5 mmol/L Ca2+、4 mmol/L Mg2+的Tris-HCl(pH 8.6)緩沖溶液。加入DNA酶 800 U,蛇毒磷酸二酯酶(Snake 酶)0.1 U,37 ℃避光孵育20 h。隨后將消化后的反應液進行固相萃取凈化富集。
固相萃取:稱取100 mg C18固相萃取吸附劑,甲醇活化并裝入固相萃取小柱,5 mL水替換甲醇,取1 mL反應液通過固相萃取小柱,4 mL水溶液淋洗,抽干小柱(不滴水即可),4 mL 40%甲醇水緩慢洗脫小柱(靠重力流下),收集洗脫液并濃縮:N2吹干,復溶于500 μL甲醇,經0.45 μm PTFE針頭濾器過濾后待測。
(±)anti-BPDE與dG反應合成BPDE-脫氧鳥苷加合物(±)anti-BPDE-N2-dG:參照王海林課題組的方法[15],稱取0.1 mg (±)anti-BPDE溶于100 μL四氫呋喃-三乙胺(19∶1,體積比)溶液中;稱取4.8 mg的2′脫氧鳥苷(dG)溶于1.2 mL 50 mmol/L Tris-HCl(pH 7.5)緩沖溶液中,將兩者混勻,避光,室溫反應48 h。
固相萃取:稱取100 mg C18固相萃取吸附劑,甲醇活化并裝入固相萃取小柱,5 mL水替換甲醇,將反應液通過固相萃取小柱,再分別用2 mL的超純水、1 mL 10%甲醇水溶液、1 mL 30%甲醇水溶液淋洗,抽干小柱(不滴水即可)后,用2 mL甲醇洗脫小柱(靠重力流下),收集洗脫液并濃縮(室溫揮發)至1 mL后待測。
1.2.2 高效液相色譜純化合成的產物經固相萃取后不能去除副產物四醇-BaP的干擾,因此,需繼續濃縮后經高效液相色譜儀(DAD檢測器)進一步純化。選用PFP色譜柱(250 mm×4.6 mm,5 μm),流動相為乙腈-0.1%甲酸水(22.5∶77.5,體積比),流速1.2 mL/min,柱溫30 ℃,進樣量20 μL,紫外檢測波長345 nm。反復多次進樣,并分別收集保留時間為22.887、28.090、30.430 、33.763 min的四種異構體流出物,經冷凍干燥后,分別復溶于500 μL的33%甲醇水溶液中,由圓二色譜儀對四種組分進行定性分析,確定四種立體異構體出峰順序。將定性后的四種立體異構體分別用紫外光譜定量,測定345 nm處紫外吸光度,根據ε345 nm=3.43×104L/(mol·cm)算出各物質濃度[18]。
1.3 HPLC-MS/MS檢測anti-BPDE-N2-dG四種立體異構體
色譜條件:PFPP色譜柱(250 mm×4.6 mm,5 μm);流動相:乙腈-0.1%甲酸水(28∶72),流速:0.5 mL/min;柱溫:30 ℃;進樣量:5 μL。
質譜條件:以正離子多反應監測(MRM)模式檢測,碰撞電壓:12 eV,母離子質荷比(m/z)為570.15,子離子m/z為454.10;采用電噴霧離子源(ESI+);霧化氣:3 L/min,離子源加熱氣:10 L/min,干燥氣:10 L/min,離子源溫度:250 ℃,脫溶劑管溫度:150 ℃;加熱塊溫度:350 ℃;碰撞誘導解離氣:270 kPa;采集時間(Dwell time):100 ms。
2 結果與討論
2.1 anti-BPDE-N2-dG四種立體異構體的制備
2.1.1 anti-BPDE-N2-dG合成方法的選擇使用HPLC-MS/MS定量檢測苯并芘DNA加合物時,由于無市售的加合物標準品,需要合成相應的純品。根據文獻報道,苯并芘的脫氧鳥苷加合物主要有兩種合成方法,一種是BPDE與小牛胸腺DNA反應生成BPDE的脫氧鳥苷酸加合物(BPDE-dGMP)[21],另一種為(±)anti-BPDE直接與2′脫氧鳥苷(dG)反應生成anti-BPDE-N2-dG[15]。本實驗對兩種方法進行了對比。第一種(具體步驟見“1.2.1”)中,BPDE與小牛胸腺DNA反應后,酶解至單核苷酸,經固相萃取純化,質譜子離子掃描[21],驗證合成BPDE-dGMP。但此合成過程中(±)anti-BPDE的用量大,因為DNA中除鳥嘌呤外,還有其他堿基會與BPDE反應從而消耗BPDE;合成過程需酶解,耗時且酶價格較昂貴,而繁瑣的反應過程也使得實驗的重復性較差。第二種方法選擇(±)anti-BPDE與dG直接反應生成(±)anti-BPDE-N2-dG,經固相萃取初步純化后,采用質譜子離子掃描[15,17]驗證合成的anti-BPDE-N2-dG。該方法中(±)anti-BPDE的用量少,從經濟和安全性角度均非常有益,且反應無需酶解,過程簡單方便,因此,本實驗選用該方法合成BPDE的DNA加合物標準品。
2.1.2 高效液相色譜純化目前,文獻報道的純化方法采用苯基柱需85 min[15]才能將anti-BPDE-N2-dG兩對對映體實現色譜分離提純,時間較長。本實驗采用常規的C18色譜柱(250 mm×4.6 mm, 5 μm)對其進行分離,結果顯示在流動相條件為乙腈-水(18.5∶81.5,體積比),流速0.75 mL/min,需要160 min才可實現六種物質(BPDE-N2-dG四種立體異構體和兩種四醇-BaP)的分離,時間很長。因此,本實驗嘗試將不同于C18柱和苯基柱作用機制的五氟苯基柱(PFP柱)應用于兩對對映體的分離,并與常規C18色譜柱進行對比,結果如圖2所示。圖中星形標記的為anti-BPDE-N2-dG立體異構體,峰1和峰2為副產物四醇-BaP的異構體。結果表明:在相同流動相條件下(乙腈-水,體積比為26∶74,流速1.2 mL/min),345 nm處紫外光譜檢測,anti-BPDE-N2-dG的四種立體異構體在C18柱上只能看到兩個峰(圖2A),而在相同規格的PFP色譜柱上(圖2B)可觀察到四種立體異構體的分離。
與中性或堿性流動相相比,酸性流動相可增強cis-(+)-anti-BPDE-N2-dG的穩定性,并提高四種立體異構體的分離度[19]。因此,在水中加入0.1%甲酸作為流動相,PFP色譜柱在相同流動相比例下(乙腈-0.1%甲酸水,體積比26∶74,流速1.2 mL/min),可觀察到anti-BPDE-N2-dG四種立體異構體中的第2、3、4峰的分離度明顯增大,且第3與第4個峰已達到基線分離(圖2D)。在此酸性流動相條件下,anti-BPDE-N2-dG的四種立體異構體在C18柱上可分離出三個峰(圖2C),較中性流動相的兩個峰(圖2A)有明顯改善,但分離效果不及PFP色譜柱(圖2D)。
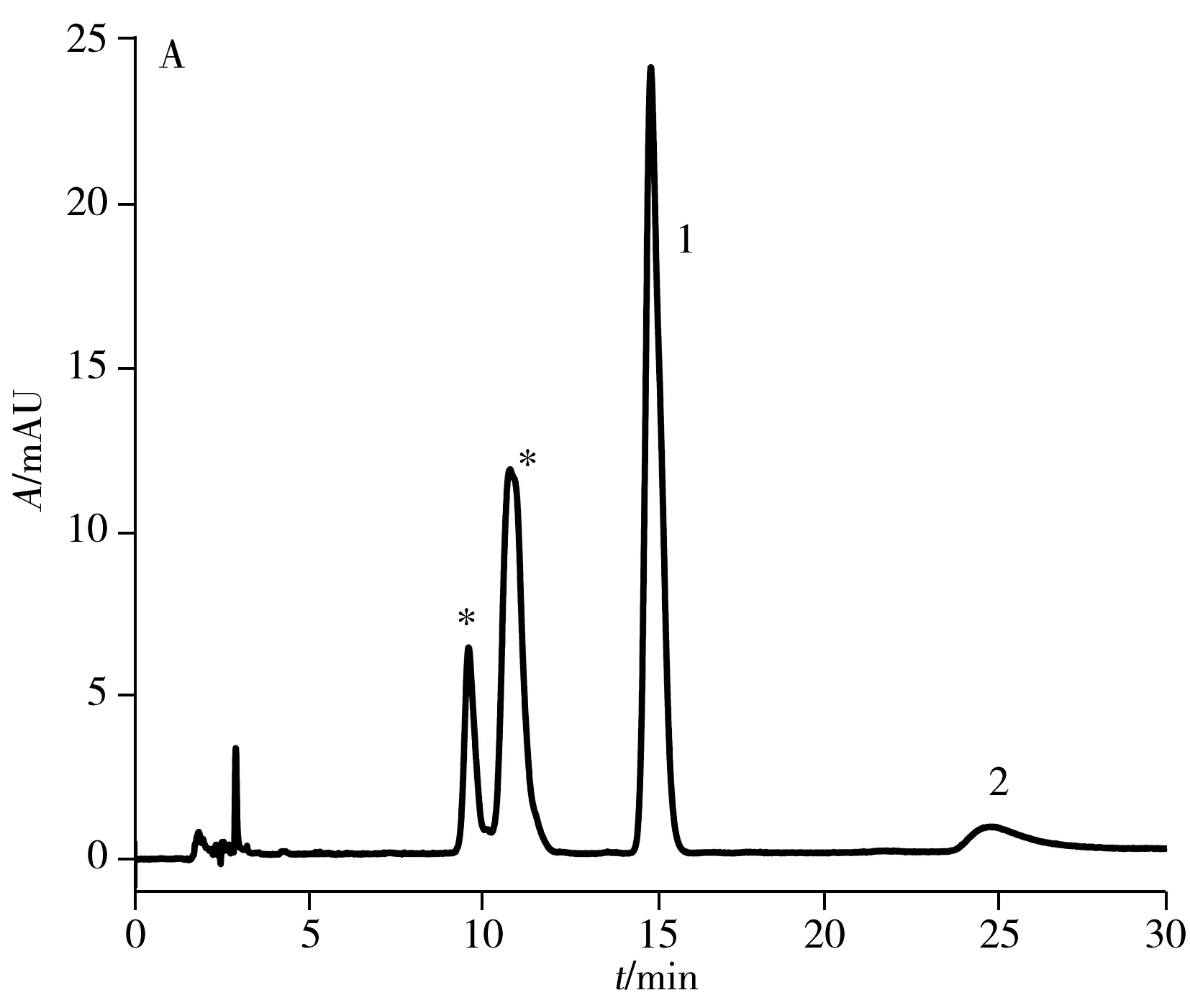
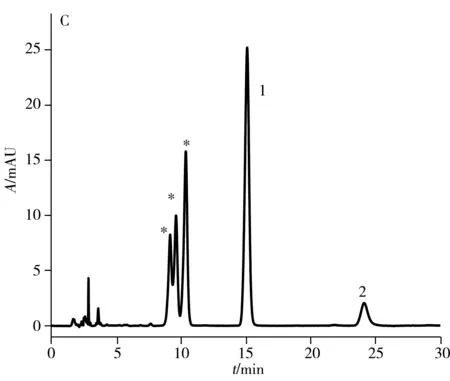
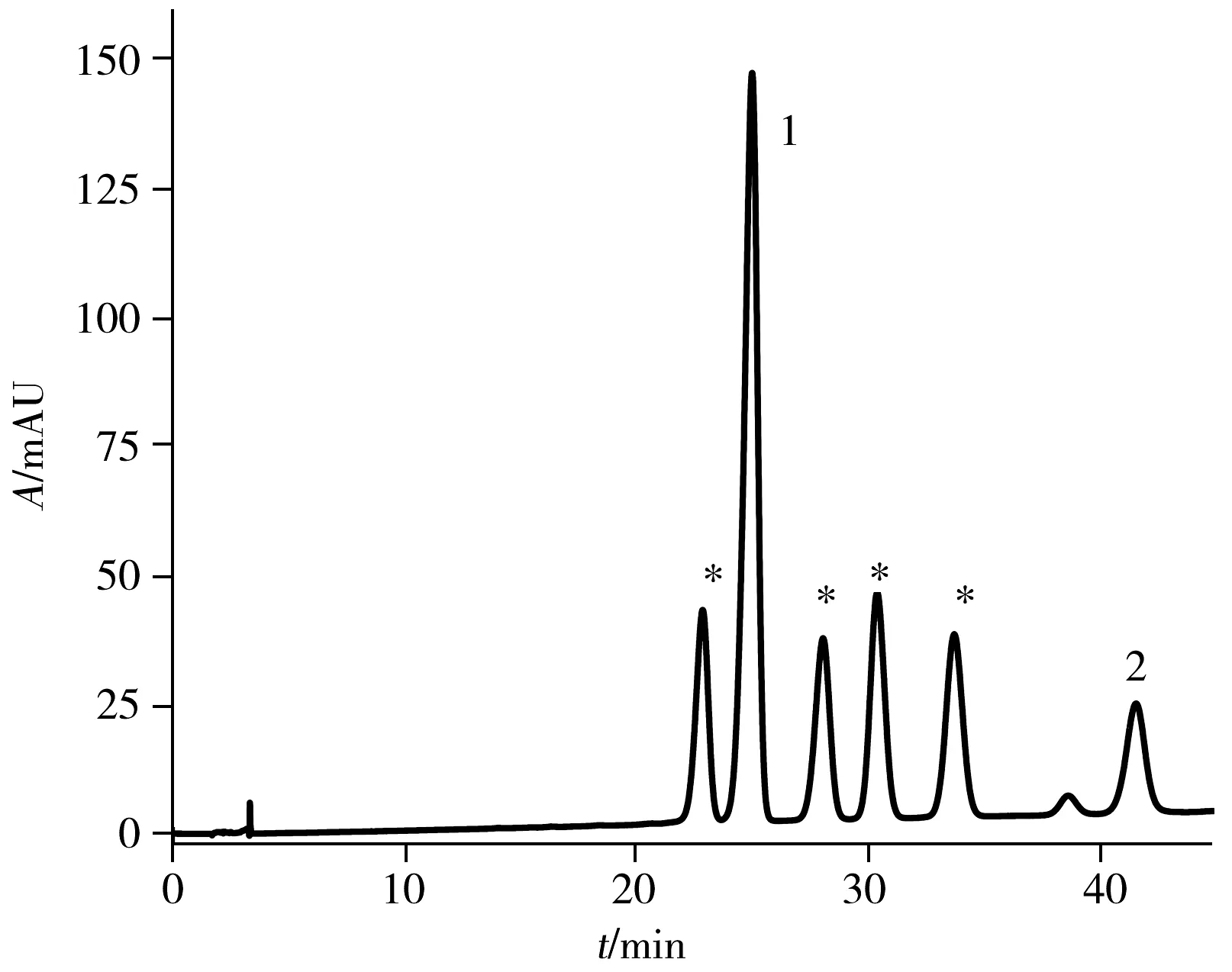
圖3 PFP色譜柱上分離anti-BPDE-N2-dG四種立體 異構體及兩種四醇-B[a]P的液相色譜圖Fig.3 HPLC chromatogram of four anti-BPDE-N2-dG stereoisomers and two BPDE tetrols obtained on PFP column the peaks of 1 and 2 are two BPDE tetrols,and the peaks labeled with asterisk are anti-BPDE-N2-dG stereoisomers;mobile phase: acetonitrile-0.1% formic acid(22.5∶77.5);λ=345 nm
為了進一步增加四種立體異構體在PFP色譜柱上的分離度,繼續調整流動相的比例,發現隨著有機相比例的減少,四種立體異構體的分離度增加,六種物質(anti-BPDE-N2-dG順反兩對異構體和兩種四醇-B[a]P)的保留時間均向后移,但anti-BPDE-N2-dG后移速度快于四醇-B[a]P。因此,anti-BPDE-N2-dG與四醇-B[a]P的峰出現重疊,流動相比例為乙腈-0.1%甲酸水(22.5∶77.5),流速1.2 mL/min時,45 min內可將六種物質完全分離開,分離度均大于2(如圖3)。分別收集anti-BPDE-N2-dG四種立體異構體,即保留時間分別為22.887、28.090、30.430、33.763 min的流出物并冷凍干燥去除溶劑,復溶于33%甲醇水溶液中進行圓二色譜(CD)及紫外吸收光譜分析。
2.1.3 anti-BPDE-N2-dG合成條件的優化為了提高anti-BPDE-N2-dG的合成產量,本實驗對(±)anti-BPDE直接與dG反應的溫度和時間進行了考察。控制其他反應條件不變,將相同的兩份樣品分別置于室溫(23.6 ℃)與37 ℃,避光反應24 h;固相萃取初步純化后,HPLC-DAD檢測(乙腈-水,26∶74,C18柱)anti-BPDE-N2-dG與四醇-B[a]P的含量(如圖2A),并比較其峰高。結果如表1所示:表中anti-BPDE-N2-dG為圖2A中保留時間為11 min的峰,四醇-B[a]P 1為圖中保留時間為15 min的峰1,四醇-B[a]P 2為圖中保留時間為25 min的峰2。經兩獨立樣本t檢驗顯示,室溫下anti-BPDE-N2-dG的產量較37 ℃高(P<0.05)(表1),而四醇-B[a]P的量下降,表明室溫下更有利于主反應的進行。控制其他反應條件不變,僅將樣品在室溫反應時間由24 h延長至48 h,結果顯示:(±)anti-BPDE與dG反應48 h時,anti-BPDE-N2-dG與四醇-BaP的峰高較24 h均提高2倍多(見表1)。為了提高anti-BPDE-N2-dG的合成產量,本實驗將反應條件定為室溫反應48 h。
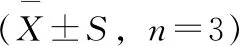

Experimental conditionAanti-BPDE-N2-dG(mAU)ABPDE tetrol 1(mAU)ABPDE tetrol 2(mAU)37 ℃,24 h52.17±0.28 74.17±0.73 2.56±0.37 23.6 ℃,24 h56.42±0.68 71.02±0.48 1.37±0.35 23.6 ℃,48 h 142.2±3.01 195.0±0.99 3.35±0.18
2.2 anti-BPDE-N2-dG立體異構體的表征及定量
在HPLC-DAD上,anti-BPDE-N2-dG與副產物四醇-B[a]P的紫外吸收光譜很相似,但仍可通過比較紫外圖譜中280~282 nm和344~347 nm處相對吸收強度來區分anti-BPDE-N2-dG與四醇-B[a]P[15]。如圖4所示,anti-BPDE-N2-dG的紫外吸收在280~282 nm處相對較高,而兩種四醇-B[a]P的紫外吸收在344 nm處相對較高。此外,根據最長波長的紫外吸收峰位置還可粗略區分順反異構體,cis(+)和cis(-)對映異構體最長的紫外吸收峰位于347 nm,而trans(+)和trans(-)對映異構體最長的紫外吸收峰位于346 nm處,cis-anti-BPDE-N2-dG相比于trans-anti-BPDE-N2-dG略有紅移[15,19]。
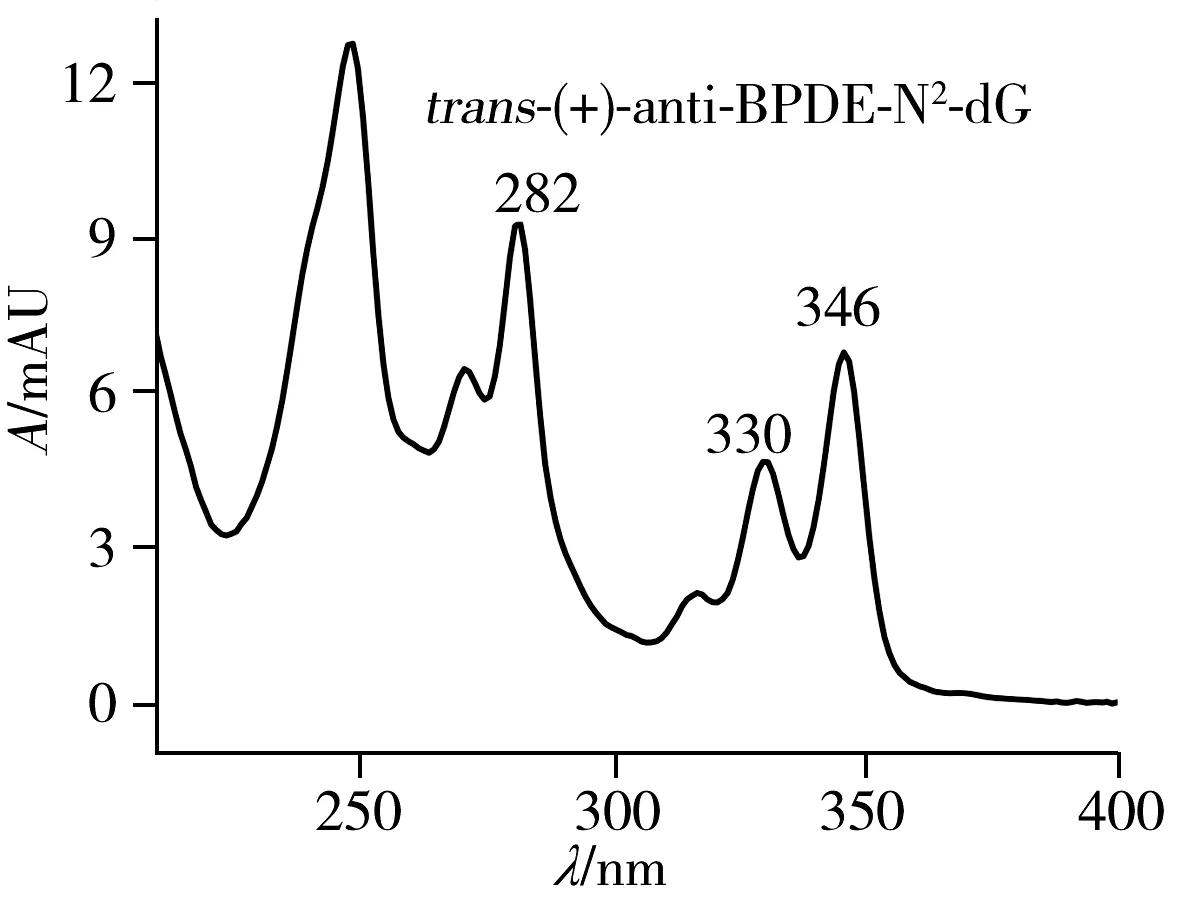
為了準確區分四種立體異構體,以及其中的對映異構體,將收集的四種組分復溶于33%甲醇水溶液中進行圓二色譜(CD)表征。對映異構體加合物trans-(+)和trans-(-),cis-(+)和cis-(-)的CD光譜相似,但符號相反。trans-(+)-和cis-(-)-anti-BPDE-N2-dG均具有10S的絕對構型,且對其最強的CD波段(250 nm)均表現出正信號。而trans-(-)-和cis-(+)-anti-BPDE-N2-dG均具有10R的絕對構型,且在相同的CD波段(250 nm)表現出負信號(圖5)。研究結果與文獻一致[19,22]。因此,確定PFP色譜柱分離anti-BPDE-N2-dG四種立體異構體的出峰順序為trans(-)(22.887 min)、trans(+)(28.090 min)、cis(+)(30.430 min)、cis(-)(33.763 min)-anti-BPDE-N2-dG(圖3)。將復溶于33%甲醇水溶液的各異構體溶液,測定其在345 nm處的紫外吸光度,可根據ε345 nm=3.43×104L/(mol·cm)算出各物質的濃度[18]。
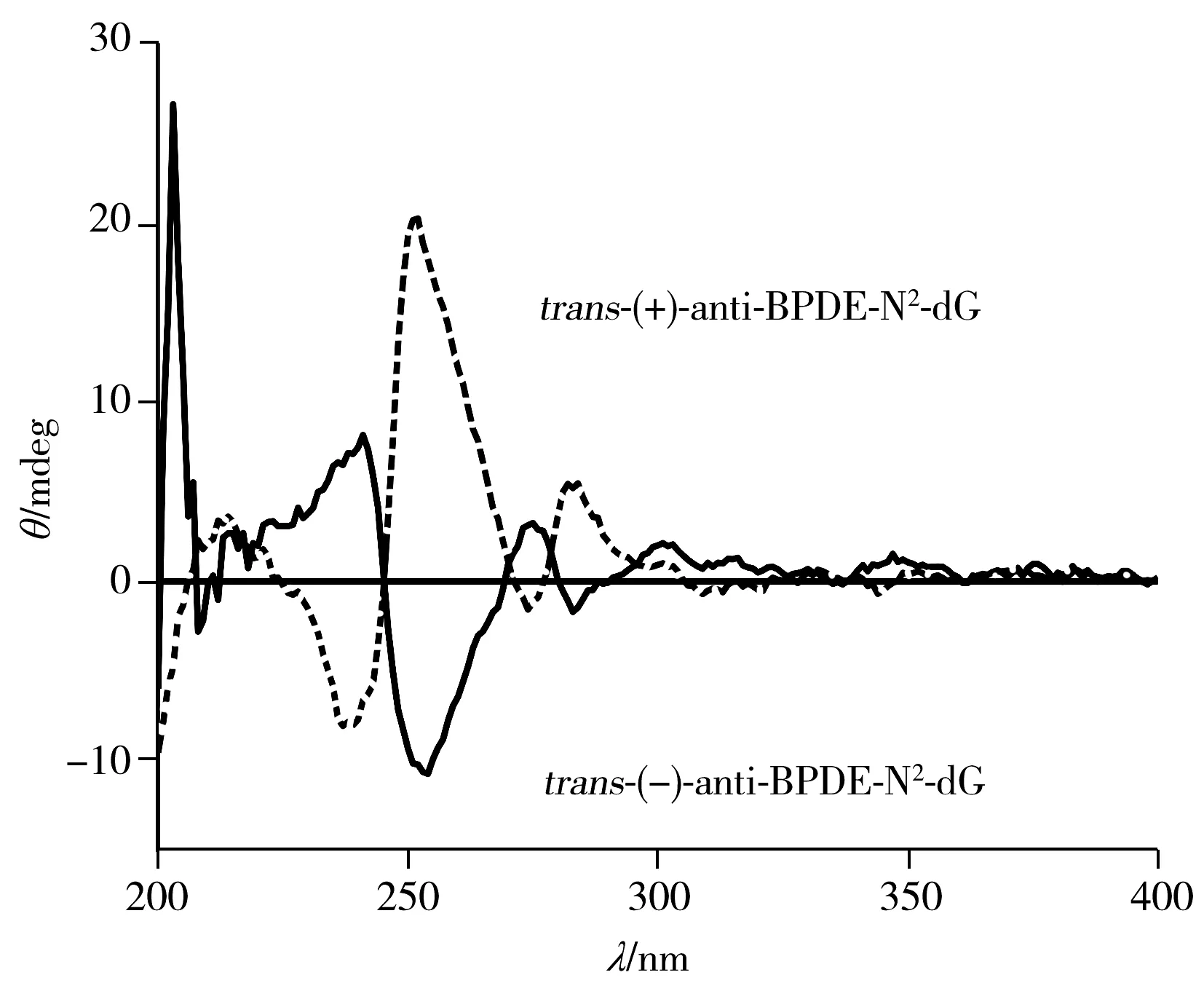

圖6 HPLC-MS/MS檢測anti-BPDE-N2-dG 四種立體異構體的色譜圖Fig.6 Chromatogram for separation of four anti-BPDE-N2-dG stereoisomers recorded by HPLC-MS/MS
2.3 HPLC-MS/MS色譜分離條件的選擇
利用HPLC-MS/MS對樣品進行檢測時,質譜在離子選擇模式下檢測可排除四醇-B[a]P的干擾,因此可不考慮四醇-B[a]P對色譜分離的影響,將anti-BPDE-N2-dG四種立體異構體分開即可滿足檢測條件。如果將HPLC-DAD分離anti-BPDE-N2-dG的條件:乙腈-0.1%甲酸水(22.5∶77.5),1.2 mL/min,PFP柱(250 mm×4.6 mm,5 μm)直接應用于HPLC-MS/MS,1.2 mL/min流速的對于質譜過高,不利于霧化,靈敏度偏低,故將流速設為0.5mL/min,調整流動相比例為乙腈-0.1%甲酸水(28∶72),柱溫30 ℃,即可在30 min內分離四種立體異構體(圖6),最小分離度可達0.995。與相同規格的苯基柱需要60 min才能將anti-BPDE-N2-dG四種立體異構體分離相比,大大縮短了分離時間,提高了分離效率,節約了分離成本。
3 結 論
本文對現有的合成純化anti-BPDE-N2-dG四種立體異構體(包含兩對對映異構體)方法進行了改進。通過優化反應溫度和時間,提高了反應產量,為定量檢測生物體苯并芘DNA加合物提供了標準品;并首次將五氟苯基色譜柱應用于anti-BPDE-N2-dG四種立體異構體的色譜分離,利用HPLC可以實現45 min內分離提純四種立體異構體,建立了一種快速純化與分離anti-BPDE-N2-dG四種立體異構體的方法。利用HPLC-MS/MS檢測四種anti-BPDE-N2-dG立體異構體標準品時,使用常規的五氟苯基色譜柱可在30 min內完成分離,大大提高了檢測效率。制備純化以及檢測分離anti-BPDE-N2-dG均可在同一色譜柱上完成,節約了分離成本。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
