分散固相萃取結合超高效液相色譜-串聯質譜法同時測定柑橘中春雷霉素與噻霉酮殘留
2020-05-08 13:40:44康霞麗趙其陽張耀海王成秋焦必寧
分析測試學報 2020年3期
關鍵詞:檢測
安 姣,康霞麗,楊 秦,趙其陽,張耀海,王成秋,焦必寧
(中國農業科學院/西南大學柑桔研究所,農業農村部柑桔產品質量安全風險評估實驗室(重慶), 農業農村部柑桔及苗木質量監督檢驗測試中心,重慶 400712)
柑橘屬作為世界第一大水果,深受人們的喜愛,但其在種植中易受到黃龍病、瘡癡病、潰瘍病等病害的侵染,雖可使用殺菌劑進行防治,但同時也會產生農藥殘留問題。春雷霉素(Kasugamycin,結構式見圖1)屬于氨基糖苷類抗生素,易溶于水,是一種環境友好的內吸性殺菌劑[1]。噻霉酮(Benziothiazolinone)屬有機雜環類殺菌劑(異噻唑啉酮類衍生物),對細菌、真菌均有較強的抑制毒殺作用。這兩種農藥均可有效防治柑橘潰瘍病、水稻稻瘟病、黃瓜細菌性角斑病、煙草野火病等,且田間藥效試驗表明,二者混劑對番茄斑疹病的防治效果顯著高于單劑[2]。因連續使用抗生素易產生抗藥性[3],春雷霉素宜與其它不同作用機制的殺菌劑交替或混合使用[1]。美國[4]、日本[5]和歐盟[6]規定春雷霉素的最大殘留限量(MRL)為0.01~0.6 mg/kg;我國GB 2763-2019[7]規定春雷霉素在柑、橘、橙上的 MRL為0.1 mg/kg,而噻霉酮在柑橘上暫無限量標準(其他作物上的MRL為0.05~1 mg/kg)。
目前,國內外春雷霉素的殘留檢測方法主要有高效液相色譜法(HPLC)[8]、反向離子對液相色譜法(IP-HPLC)[9-11]、液相色譜串聯質譜法(LC-MS/MS)[12-18]和高效毛細管電泳法[19]、膠束液相色譜法[20]、生物測定法[21]等,樣品多為水、土壤、蔬菜、谷物等。春雷霉素極性大且無發色基團,在反向色譜柱上難保留,分析檢測難度較大。雖然可通過衍生化操作引進發色基團進行紫外檢測,但該過程操作繁瑣且易引入未知雜質[22]。Sheu等[8]利用高效液相色譜(HPLC/UV)檢測了水中的春雷霉素含量,但靈敏度較低(定量下限為2.2 mg/L)。牛長群、吳國旭等[11]建立了春雷霉素的反相離子對高效液相色譜檢測方法,通過加入離子對試劑改變保留時間;但該法對于試劑、濃度、室溫有較高要求,且保留時間過長(14.5 min),不適合大量樣品快速檢測。液相色譜串聯質譜法選擇性好,定性定量能力強,但凈化方式基本以固相萃取(SPE)為主,迄今尚未見分散固相萃取(DSPE)凈化春雷霉素的相關文獻報道。噻霉酮殘留的檢測方法較少,且主要集中在材料、化妝品、涂料等工業領域,其在食品中的殘留檢測主要以液相色譜法[23-24]為主,粟有志等[25]采用QuEChERS前處理方法結合 HPLC-MS/MS測定了果蔬中的噻霉酮。由于上述兩種農藥均無國家及行業標準的檢測方法,也未見到柑橘中相關檢測方法報道,因此建立柑橘中春雷霉素和噻霉酮的檢測方法具有重要意義。
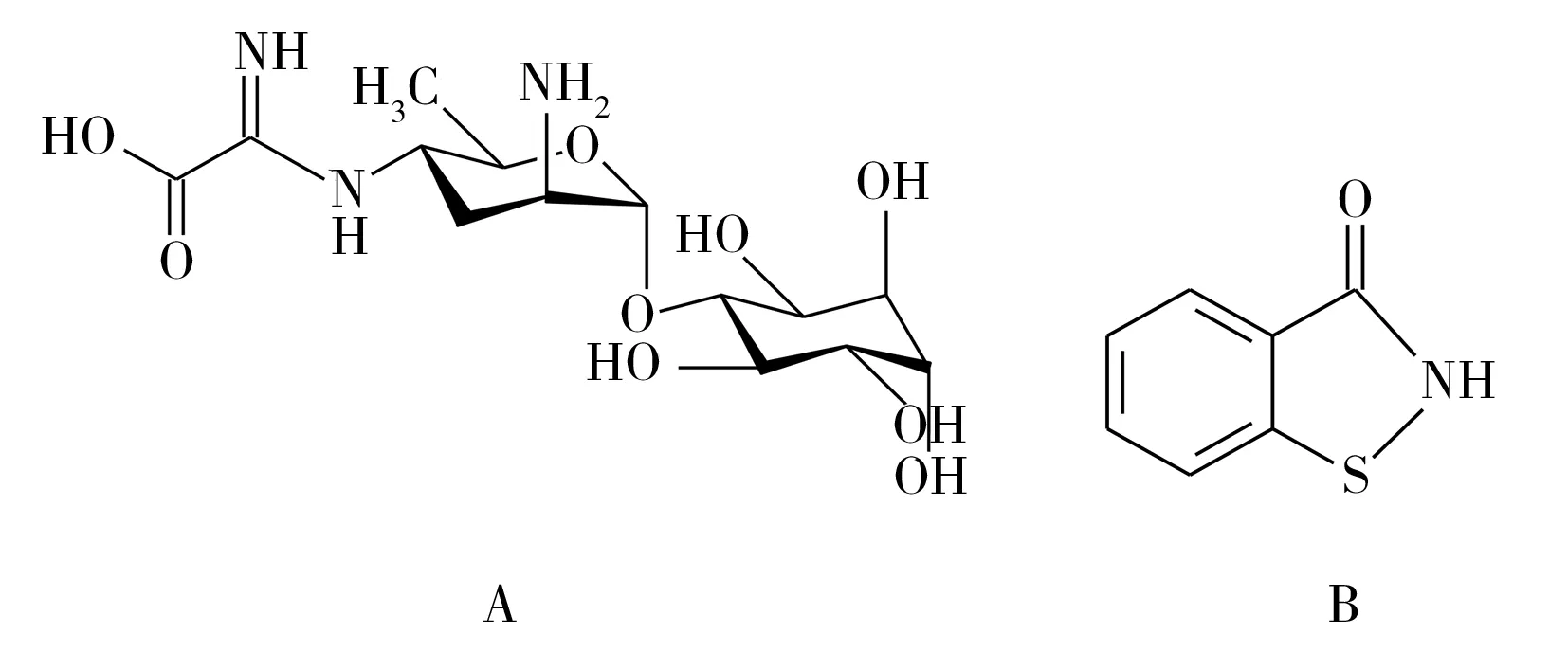
圖1 春雷霉素(A)和噻霉酮(B)結構式Fig.1 Structural formula of kasugamycin (A) and benziothiazolinone (B)
本研究采用分散固相萃取法凈化樣品,結合超高效液相色譜-串聯質譜法(UPLC-MS/MS),建立了同時測定柑橘中春雷霉素和噻霉酮殘留的分析方法,該方法靈敏度高,準確度好,方便快捷,能夠滿足分析檢測要求,填補了柑橘中春雷霉素和噻霉酮同時檢測的空白,并為噻霉酮在柑橘上的限量標準制訂提供了數據支持,為食品中農藥殘留安全性評價提供了科學依據。
1 實驗部分
1.1 儀器與試劑
Nexera X2 超高效液相色譜儀(日本Shimadzu公司),QTRAP?6500三重四極桿/復合線性離子阱質譜(美國AB公司),IonDriveTMTurbo V 離子源,Analyst?1.6.3工作站;CL31/CL31R 多用途離心機(美國Thermo Fisher公司);Milli-Q A10超純水儀(美國Millipore公司);KQ5200DE型數控超聲波清洗器(昆山市超聲儀器有限公司);CK2000型高通量組織研磨儀(北京Thmorgan生物科技有限公司)。
春雷霉素(89.0%)、噻霉酮(99.6%)(德國 Dr.Ehrenstorfer公司);乙腈、甲醇(色譜純,德國 CNW公司);N-丙基乙二胺(PSA)、超純反相C18填料(加拿大 SiliCycle公司);石墨化炭黑(GCB,德國 CNW公司)。
1.2 標準溶液配制與標準曲線的繪制
用甲醇分別配制質量濃度1 000 mg/L的春雷霉素和噻霉酮單標儲備液,分別移取適量標準儲備液用甲醇稀釋成100 mg/L的標準混合液,-50 ℃保存。
分別用溶劑和空白基質提取液稀釋標準溶液,配制成質量濃度為0.5、1、2、5、10、50、100、200 μg/L的溶劑標準工作液和基質匹配標準工作液,按照“1.4”儀器條件進行測定,以目標物的質量濃度(μg/L)為橫坐標(x),對應峰面積值為縱坐標(y)繪制標準曲線。
1.3 前處理方法
準確稱取勻漿后的柑橘樣品2.00 g(精確至0.01 g)于50 mL聚四氟乙烯離心管中,全果樣品中加入含0.5%甲酸的乙腈-水(7∶3,體積比,下同)溶液,果肉樣品中加入含0.5%氨水的乙腈-水(7∶3)溶液,定容至20 mL,渦旋混勻,垂直振蕩30 min后,10 000 r/min 離心5 min。移取2 mL上清液置于裝有50 mg C18的離心管中,渦旋1 min,3 000 r/min 離心5 min,取上清液經0.22 μm 有機濾膜過濾,待測。
1.4 儀器條件
1.4.1 色譜條件Waters ACQUITY UPLC?HSS T3 色譜柱(150 mm×2.1 mm,1.8 μm);流動相:A為0.2%甲酸水溶液,B為甲醇。梯度洗脫程序:0~2 min,2% B,2~4 min,2%~98% B,4~8 min,98% B,8~8.1 min,98%~2% B,8.1~9 min,2% B。柱溫40 ℃;進樣量5 μL;流速0.2 mL/min。
1.4.2 質譜條件電噴霧離子源正離子模式(ESI+);多反應離子監測(MRM);氣簾氣(CUR):20 psi;離子化電壓(IS):4 500 V;電噴霧離子源溫度(TEM):500 ℃;噴霧氣:50 psi;輔助加熱氣:50 psi;碰撞室入口電壓(EP):10 V;碰撞室出口電壓(CXP):10.5 V。其余質譜參數見表1。

表1 春雷霉素和噻霉酮的質譜參數Table 1 MS/MS parameters for kasugamycin and benziothiazolinone
*quantification ion
2 結果與討論
2.1 儀器條件的優化
配制0.5 mg/L 的標準溶液,以針泵恒流進樣分別在ESI+模式和ESI-模式下進行掃描,選擇效果較好的正離子模式,得到母離子為[M+H]+,進行子離子掃描,找到合適的子離子后建立MRM離子對通道,進一步優化獲得最佳去簇電壓(DP)和碰撞能量(CE)。
針對高極性農藥,有文獻使用不同色譜柱包括反向柱[26]、陰離子交換柱[27]、反相和弱陰離子交換混合模式柱[28]、親水相互作用柱等進行分析。春雷霉素是高親水化合物,其結構中含有氨基,在反向柱(如C18柱)上難保留[15],常選擇反相離子對液相色譜法(IP-HPLC)或親水相互作用(HILIC)柱進行分析,但離子對試劑會影響質譜的靈敏度,而HILIC柱更宜與ESI-MS結合使用[22]。T3色譜柱以C18鍵合相為固定相,可用純水作流動相,對極性化合物保留效果較好。本文比較了ACQUITY UPLC?HSS T3 色譜柱(150 mm×2.1 mm,1.8 μm),ACQUITY UPLC?HSS T3 色譜柱(50 mm×2.1 mm,1.8 μm)和ACQUITY UPLC?BEH HILIC色譜柱(50 mm×2.1 mm,1.7 μm)的分析效果,,發現HILIC柱上春雷霉素和噻霉酮出峰時間較為接近,兩峰有重疊;50 mm的T3柱上春雷霉素保留時間較短,無法與雜質較好分離,150 mm的T3柱上可獲得良好的峰形和分離度。故選擇150 mm 的ACQUITY UPLC?HSS T3柱。
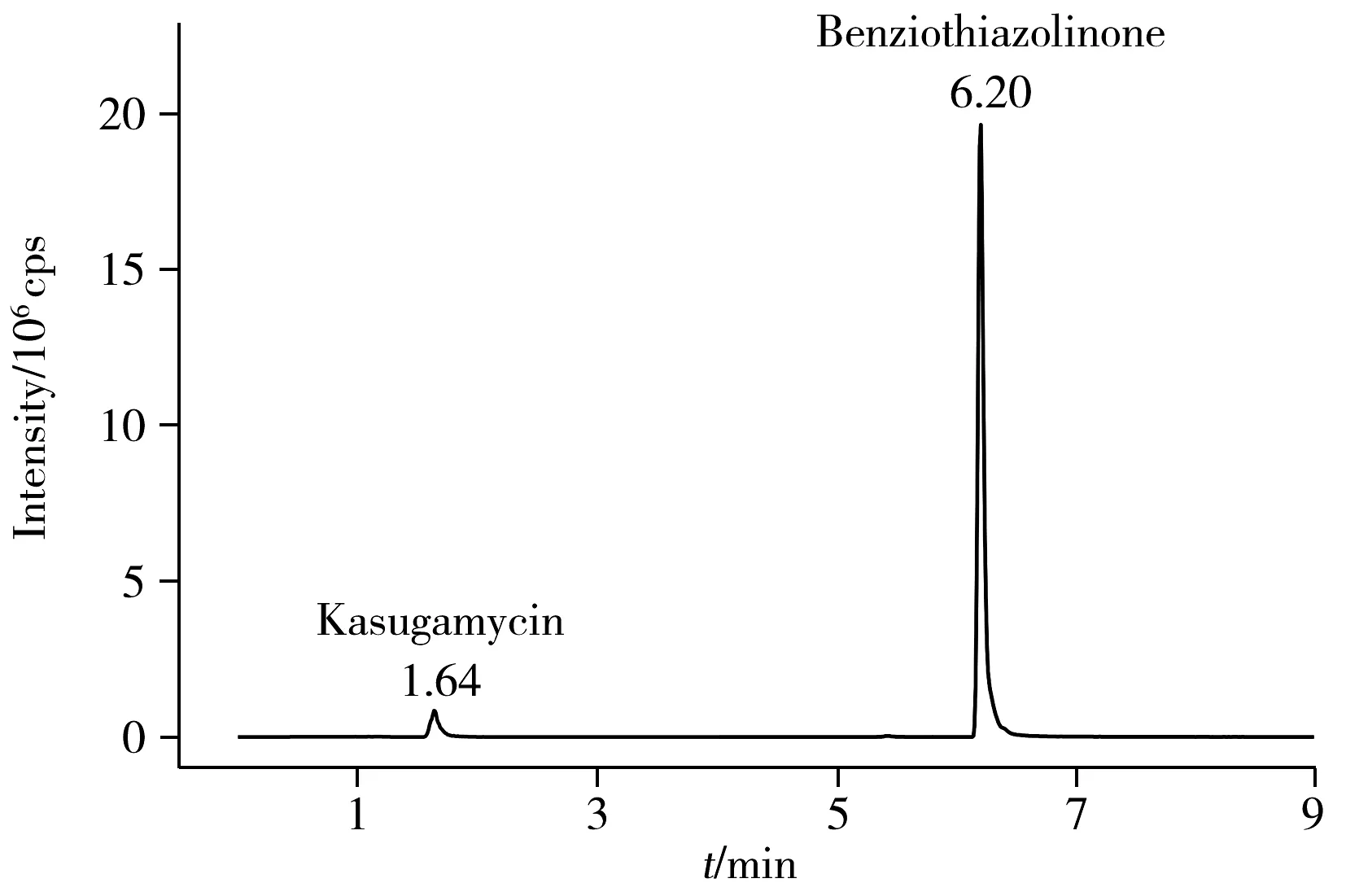
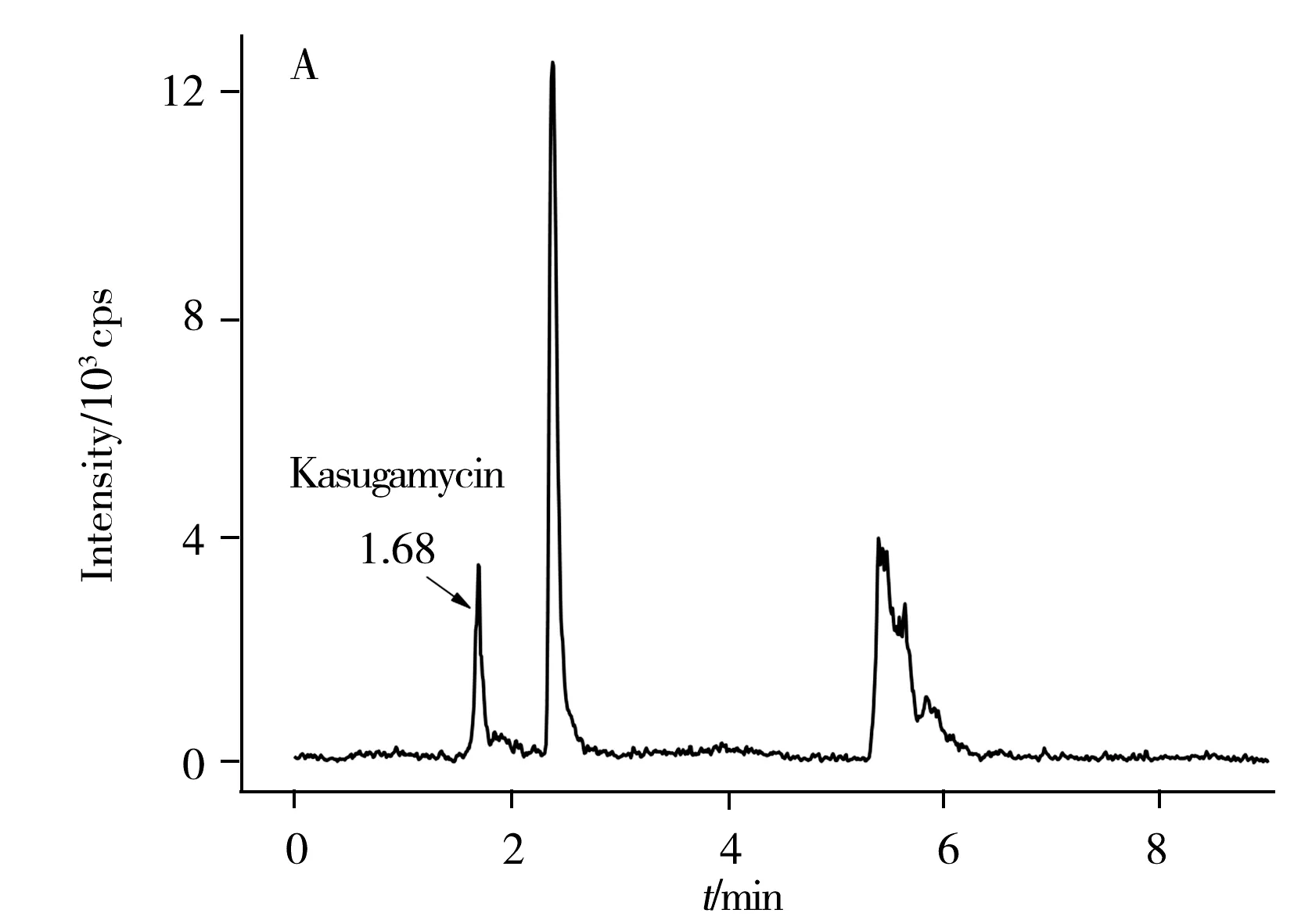
圖2 0.2 mg/L春雷霉素和噻霉酮標準溶液色譜圖Fig.2 0.2 mg/L kasugamycin and benziothiazolinone standard solution chromatogram
此外,分別比較了有機相為甲醇或乙腈,水相中加入不同含量甲酸(0.1%、0.2%)及乙酸銨時對峰形和響應值的影響。當有機相為乙腈時,噻霉酮有拖尾小峰出現。水相中加入甲酸可提高離子化效率,使響應值增加,峰形對稱,0.2%甲酸較0.1%甲酸效果好;乙酸銨可促進離子化,調節pH值,改善峰形,但加入乙酸銨會使噪音值變大。綜上所述,選擇0.2%甲酸水-甲醇體系作為流動相。在該儀器條件下的標準溶液色譜圖如圖2所示。
2.2 前處理條件的優化
2.2.1 提取劑以柑橘樣品為研究對象,考察了不同提取劑對兩種農藥的提取效果。春雷霉素和噻霉酮的提取劑多為甲醇或乙腈等極性溶劑,但試驗過程中,當提取劑為甲醇或乙腈時,春雷霉素的回收率均無法滿足要求(低于50%),而以乙腈提取噻霉酮時回收率較甲醇高,使用相同比例的甲醇-水和乙腈-水時,乙腈-水的回收率較好,故比較了不同比例的乙腈-水對全果基質的提取效果(加標水平0.1 mg/kg)。結果表明,隨著乙腈含量的增加,噻霉酮的回收率增加,但春雷霉素在乙腈體積分數為80%時回收率驟降,當乙腈-水的體積比為7∶3時,兩種目標物的回收率最好。
由于pH值對目標物的提取效率影響很大,故考察了酸堿度對回收率的影響(圖3):對于全果基質,加入0.5%甲酸時,提取效果較好;而對于果肉基質,選擇加入0.5%氨水時,目標物均滿足回收率要求。最終選擇含0.5%甲酸的乙腈-水溶液(7∶3)為柑橘全果提取劑,含0.5%氨水的乙腈-水溶液(7∶3)為柑橘果肉提取劑。
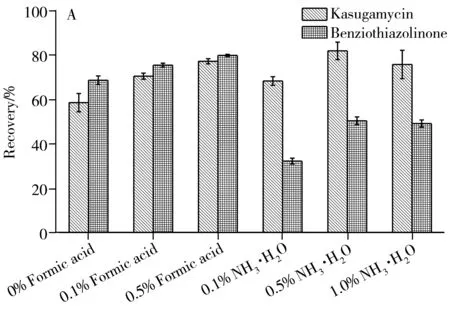
2.2.2 提取劑體積以柑橘全果為研究對象,比較了不同體積(10、20、30、40 mL)的提取劑對兩種目標物的提取效果。結果顯示,提取劑為20 mL和30 mL時,均能滿足回收率要求,考慮到檢測靈敏度和溶劑消耗,選擇20 mL作為提取劑體積。
2.2.3 提取方式以全果基質為研究對象,比較了超聲提取和振蕩提取對提取率的影響。當提取時間為30 min時,振蕩提取的提取效果較好,噻霉酮回收率較高,春雷霉素無太大變化。這可能是由于噻霉酮在水中的溶解度隨溫度上升而增大,而超聲過程產熱,噻霉酮進入乙腈層的比例減小,導致回收率較低。因此選用振蕩提取方式。
2.2.4 凈化劑在分散固相萃取(DSPE)中,目前使用最多的凈化材料是N-丙基乙二胺(PSA)、超純反相C18填料和石墨化炭黑(GCB)。PSA是一種弱陰離子交換劑,可有效去除有機酸、色素及一些糖類物質;C18可去除基質中的脂肪和脂類等非極性有機物;GCB能很好地去除色素和固醇類化合物[29]。
因此,比較了50 mg C18、PSA、GCB對于柑橘全果基質的凈化效果,凈化后,提取液顏色由淺到深依次為GCB>C18>PSA,證明GCB對色素具有良好吸附作用。當凈化劑為GCB時,噻霉酮的回收率偏低,可能由于GCB對極性分子具有強親和力,可與具有苯環等平面結構的噻霉酮發生疏水相互作用,導致凈化過程中分析物損失[30],回收率下降。當凈化劑為C18和PSA時,兩種目標物回收率均可滿足要求,其中C18略優,因此選用C18作為凈化劑。進一步考察了25、50、75、100 mg C18對回收率的影響,結果表明:C18用量為25~75 mg時,春雷霉素回收率逐步上升,噻霉酮則在用量為50 mg時回收率較好,故最終選擇50 mg C18作為凈化劑。
2.3 標準曲線與檢出限
樣品本身的內源性組分和前處理引入的外源性組分產生的基質效應影響方法的線性、靈敏度、精密度等[32],本法采用基質匹配標準曲線校正基質效應(ME=基質匹配標準曲線斜率/溶劑標準曲線斜率)。結果如表2所示,基質對兩種目標物均存在抑制作用,且對春雷霉素抑制作用強烈。以3倍信噪比(S/N)對應的檢測濃度確定檢出限(LOD),以最小加標濃度確定定量下限(LOQ)。在0.5~200 μg/L質量濃度范圍內,春雷霉素和噻霉酮線性良好(r2>0.999),檢出限(LODs)為0.006~0.04 μg/kg,定量下限(LOQs)為5~10 μg/kg。

表2 春雷霉素和噻霉酮的線性范圍、線性方程、相關系數、基質效應、方法檢出限和定量下限Table 2 Linear ranges,regression equations,correlation coefficients(r2),matrix effects,limits of detection(LODs) and limits of quantification(LOQs) for kasugamycin and benziothiazolinone
2.4 準確度與精密度
在柑橘全果和果肉空白基質中添加不同濃度的標準溶液,按照“1.3”方法處理,每個加標水平做6個平行。由表3可知,在5~1 000 μg/kg加標水平下,春雷霉素的回收率為79.7%~104%,相對標準偏差(RSD)為5.6%~9.6%;噻霉酮的回收率為73.4%~88.5%,RSD為1.6%~8.0%。結果表明該方法具有良好的準確度和精密度,可滿足農殘檢測分析要求。
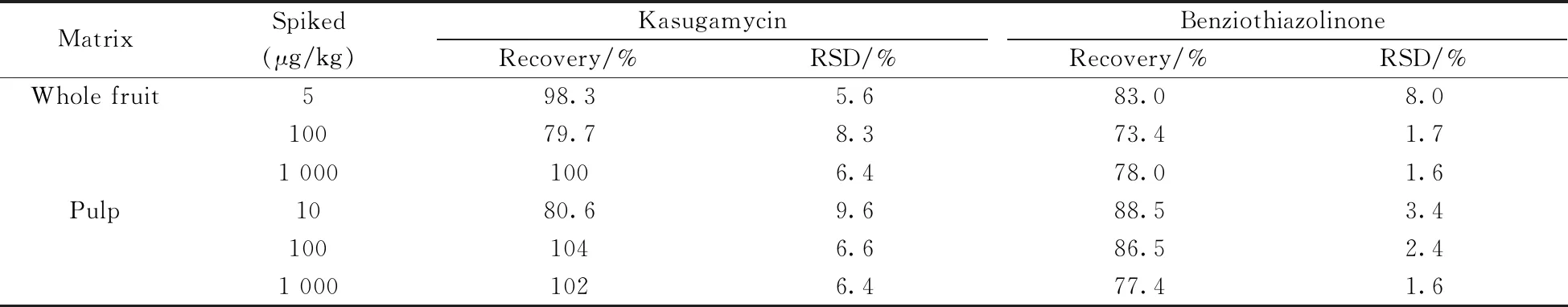
表3 不同基質中春雷霉素和噻霉酮的加標回收率和相對標準偏差(n=6)Table 3 Recoveries and relative standard deviations(RSDs) of kasugamycin and benziothiazolinone in different matrixes(n=6)
2.5 方法對比
本方法使用C18進行分散固相萃取凈化,與固相萃取小柱相比,成本低,耗時少,摒棄了丙酮等不適合液質的提取劑,簡化了前處理步驟,且回收率較好,適合大量樣品的快速檢測(表4)。
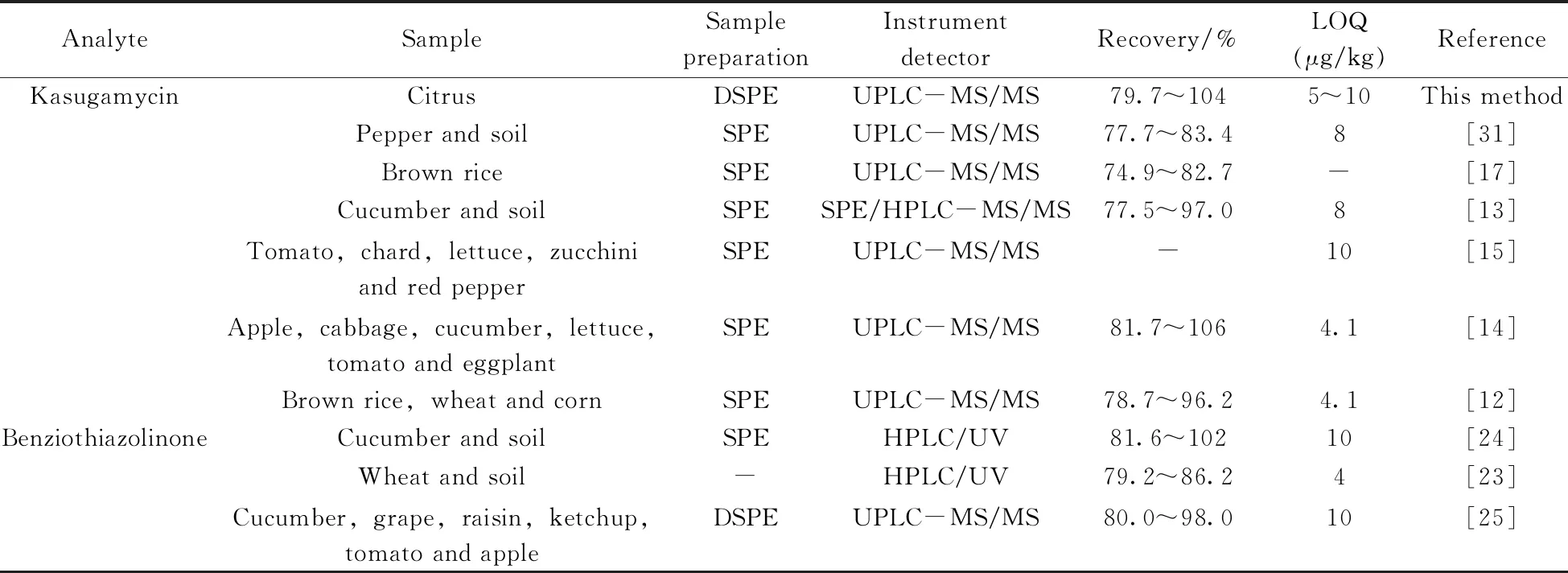
表4 春雷霉素和噻霉酮在食品中的檢測方法對比Table 4 Comparison of detection methods of kasugamycin and benziothiazolinon in food
2.6 實際樣品檢測
于2018~2019年在重慶市北碚區開展田間試驗,供試藥劑為8%春雷霉素·噻霉酮水分散粒劑,供試作物為溫州蜜柑,分別按照400 mg a.i./kg(5倍推薦劑量)的有效劑量施藥1次(末次施藥后0、1、2、3、4、5、7、14、21、28 d采集田間樣品),80 mg a.i./kg有效劑量施藥2次,施藥間隔期7 d(末次施藥后21、28 d采集田間樣品),使用本方法進行測定。根據測定結果得到:春雷霉素在溫州蜜柑中的消解符合一級動力學方程,殘留量隨時間延長逐漸減小,降解方程為y=0.601 7e-0.052x(r2=0.748 1),y=0.401 7e-0.055x(r2=0.733 3),y=0.365 6e-0.049x(r2=0.634 0),半衰期T1/2=12.60~14.15 d;噻霉酮的消解不遵循一級動力學方程,呈上升下降連續波動趨勢。由于只在單一品種和地區進行實驗,數據僅供參考,后期仍需佐證。結果顯示,全果中春雷霉素的消解殘留量為0.096 1~0.752 8 mg/kg(圖4A),噻霉酮為0.005 4~0.016 9 mg/kg(圖4B)。全果和果肉中的最終殘留量均為未檢出。
3 結 論
本文建立了DSPE/UPLC-MS/MS同時測定柑橘中水溶性春雷霉素和脂溶性噻霉酮的分析方法。使用T3色譜柱得到了良好的峰形及分離度,通過分散固相萃取法凈化,簡化了步驟并保證了良好的回收率。該方法靈敏度高,方便快捷,精密度好,能滿足柑橘水果中春雷霉素和噻霉酮殘留的檢測要求,可為制定春雷霉素和噻霉酮復配農藥的安全使用規則和在柑橘上的產品登記提供技術支撐。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
