分子印跡電化學傳感器檢測水楊酸
2020-05-08 13:40:44張夏紅林水東盧曉春李錦輝
分析測試學報 2020年3期
董 雁,張夏紅,林水東,盧曉春,李錦輝
(1.龍巖學院 化學與材料學院,福建 龍巖 364012;2.固體廢棄物資源化利用福建省高校 工程研究中心,福建 龍巖 364012)
分子印跡技術(Molecular impinting technology)是一種使聚合物聚合后與模板分子的空間構型相匹配的“空穴”結構的技術。所形成的“空穴”對模板分子有高度識別性,不易受外界干擾,穩定性與重現性好,可用于低濃度樣品的檢測[1]。分子印跡技術最早出現于生物化學,源于抗體對某種分子具有專一性識別功能的一種分析技術。之后,經過科學家的不斷探索和完善,逐漸形成了一門新興技術,得到了人們的普遍認可[2]。近年來,分子印跡技術在傳感器領域的發展更可謂是動力十足,已成為該領域最具發展潛力的先進技術之一[3-5]。將電化學聚合法與分子印跡法相結合制備印跡聚合物具有使用儀器簡單[6]、成本低、易操作等特點,且電化學聚合法制備的印跡膜厚度可控、成膜均勻、重復性較好,較化學聚合有更多優勢。但該方法也存在一些不足,如未經修飾的玻碳電極表面靈敏度不高等。通常通過引入功能性納米材料(如石墨烯等)以改善此類問題[7]。
水楊酸(SA)是一種脂溶性有機酸,為阿司匹林以及很多止痛藥的制備原料,并被廣泛用于化妝品中。但由于高濃度的SA會對皮膚造成損害,我國對化妝品中SA含量有明確規定,因此檢測其在化妝品中的含量就尤為重要。傳統檢測SA的方法有熒光分光光度法[8]、氣相色譜-質譜聯用法[9]、高效液相色譜法[10-11]等。此外,也有基于印跡聚合技術檢測SA的研究,但采用電聚合法制備SA分子印跡膜,用于檢測SA含量卻鮮見報道[12-15]。本文以鄰苯二胺和吡咯為復合功能單體,SA作為模板分子,通過循環伏安法(CV)在石墨烯修飾的玻碳電極表面制備高選擇性及高靈敏度的SA分子印跡膜。優化了分子印跡電化學傳感器的制備條件,對傳感器性能進行了表征,并采用傳感器實現了對SA的檢測。
1 實驗部分
1.1 試劑與儀器
水楊酸(SA)、鄰苯二胺(o-PPD)、乙腈(MeCN)、磷酸二氫鈉、磷酸氫二鈉、鐵氰化鉀、氯化鉀、乙酸、乙酰水楊酸、對羥基苯甲酸、苯甲酸、吡咯(Py)、苯甲醛(分析純,麥克林公司);0.5 mg/mL氧化石墨烯分散液(GO,南京先豐納米材料科技有限公司)。
玻碳電極(GCE)、CHI660C電化學工作站(上海辰華儀器公司);KQ-100DE超聲波清洗儀(昆山超聲儀器有限公司);PH-3CB/3CU型PH計(上海越平科學儀器有限公司);S-3400N掃描電子顯微鏡(日本日立公司)。
1.2 石墨烯修飾玻碳電極的制備(GO/GCE)
先將玻碳電極分別用0.5 μm和0.05 μm的Al2O3粉末對電極表面拋光處理,然后用超純水清洗3次,采用循環伏安法使其在鐵氰化鉀探針溶液中出峰電位差為85 mV后,自然風干。取5 μL氧化石墨烯分散液涂覆于預處理過的玻碳電極表面,自然晾干后再取3 μL滴涂至玻碳電極表面,自然晾干即制成石墨烯修飾玻碳電極(GO/GCE)[16]。
1.3 SA印跡電極的制備(MIP/GO/GCE)
在41 mL pH 6.6的磷酸緩沖溶液(PBS)中加入1 mL 0.1 mol/L SA溶液、4 mL 0.1 mol/Lo-PPD和4 mL 0.1 mol/L的Py溶液,充分混勻后待用。將“1.2”處理好的玻碳電極、參比電極及輔助電極所組成三電極體系浸入上述聚合液中,用循環伏安法(CV)掃描15圈,掃描范圍為-0.2~0.8 V,掃描速度為50 mV/s進行制備。聚合結束后,先用超純水反復沖洗電極,再放入洗脫液中洗脫20 min除去模板分子,自然晾干,得到SA分子印跡傳感器(MIP/GO/GCE)。非印跡分子傳感器(NIP/GO/GCE)的制備除不加SA外,其它條件保持不變。
2 結果與討論
2.1 分子印跡聚合膜的電化學行為
采用CV法分別對NIP/GO/GCE、MIP/GCE及MIP/GO/GCE的電化學行為進行考察(圖1)。結果顯示,對NIP/GO/GCE進行第1次掃描時,在0.065 V處出現氧化峰,第2圈掃描后,電流強度明顯降低,并隨著掃描圈數的增多,電流恢復到背景值(圖1A),表明o-PPD和Py可在此電流范圍內發生聚合反應,在玻碳電極表面形成不導電的致密聚合膜,致使o-PPD和Py未進一步氧化,抑制了伏安電流響應。圖1B 為MIP/GCE的印跡聚合行為,可觀察到未加石墨烯時,修飾電極聚合的氧化還原峰不明顯。而圖1C為MIP/GO/GCE電聚合過程的CV圖,可觀察到強氧化還原峰,表明石墨烯具有良好的導電性,修飾電極后能提供給反應分子較大的比表面積,使反應更容易進行。

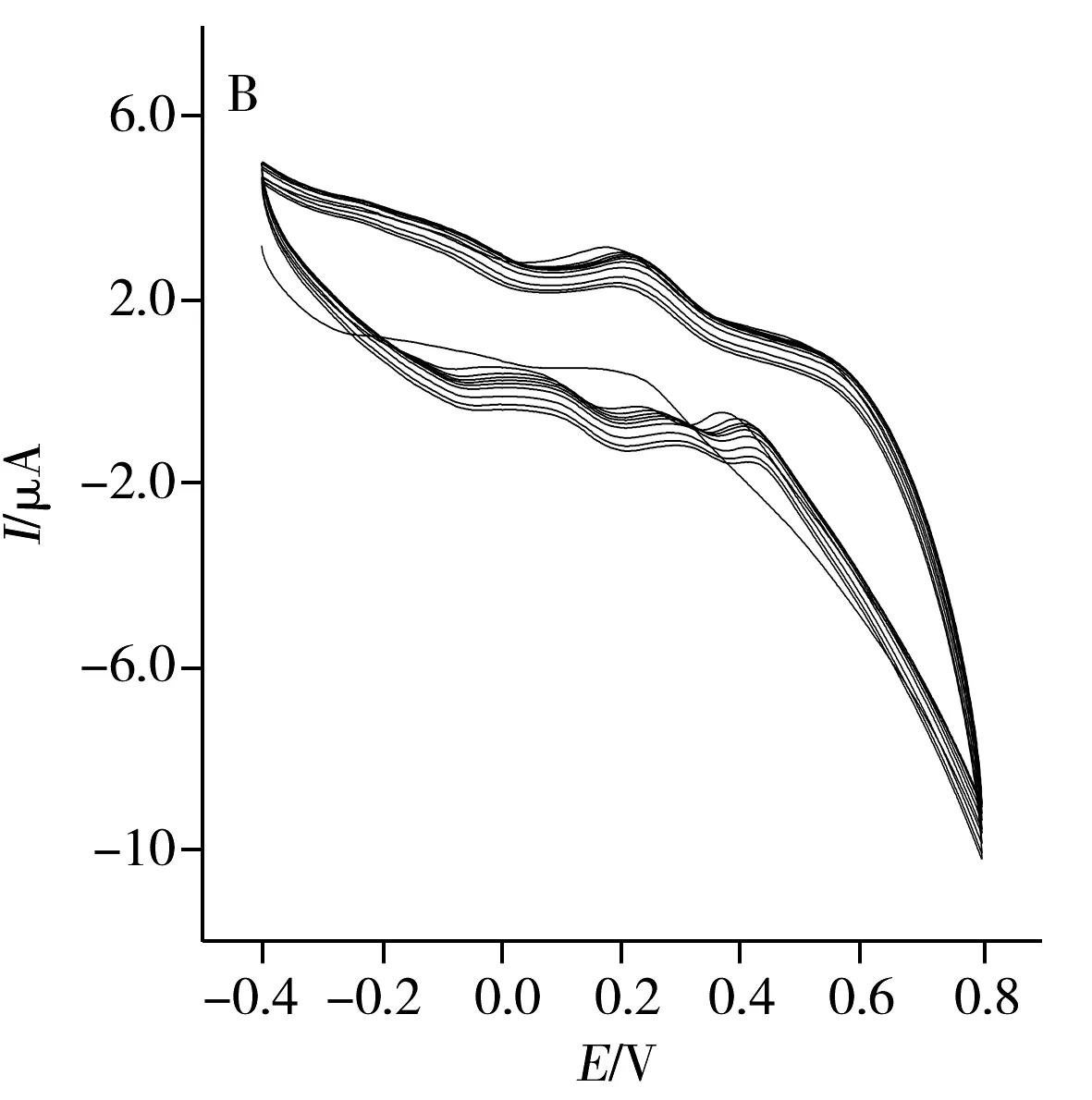
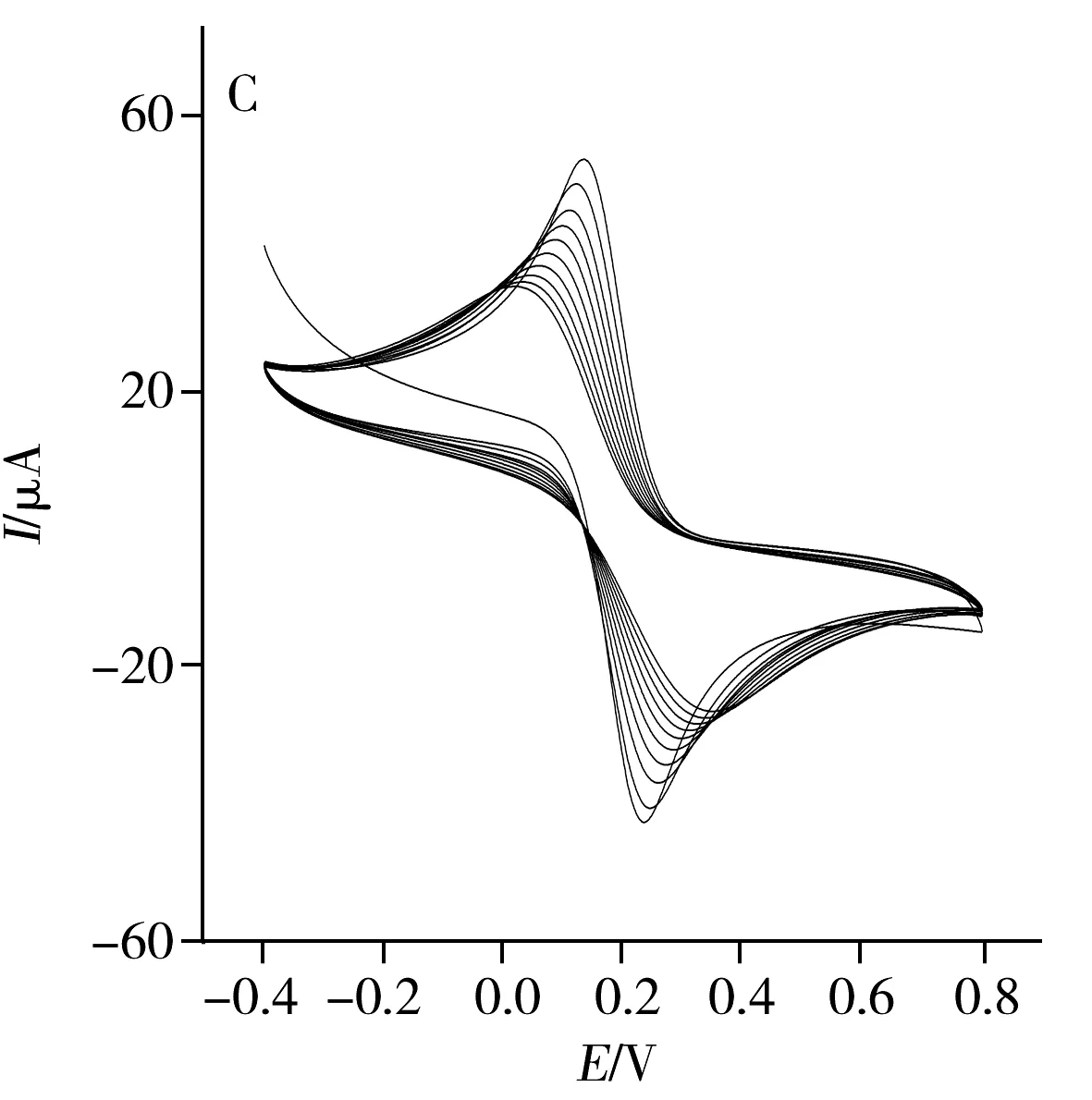
圖1 NIP/GO/GCE(A)、MIP/GCE(B)及MIP/GO/GCE(C)的CV圖Fig.1 CV curves of NIP/GO/GCE(A),MIP/GCE(B) and MIP/GO/GCE(C)
2.2 裸電極及石墨烯修飾電極的電化學表征
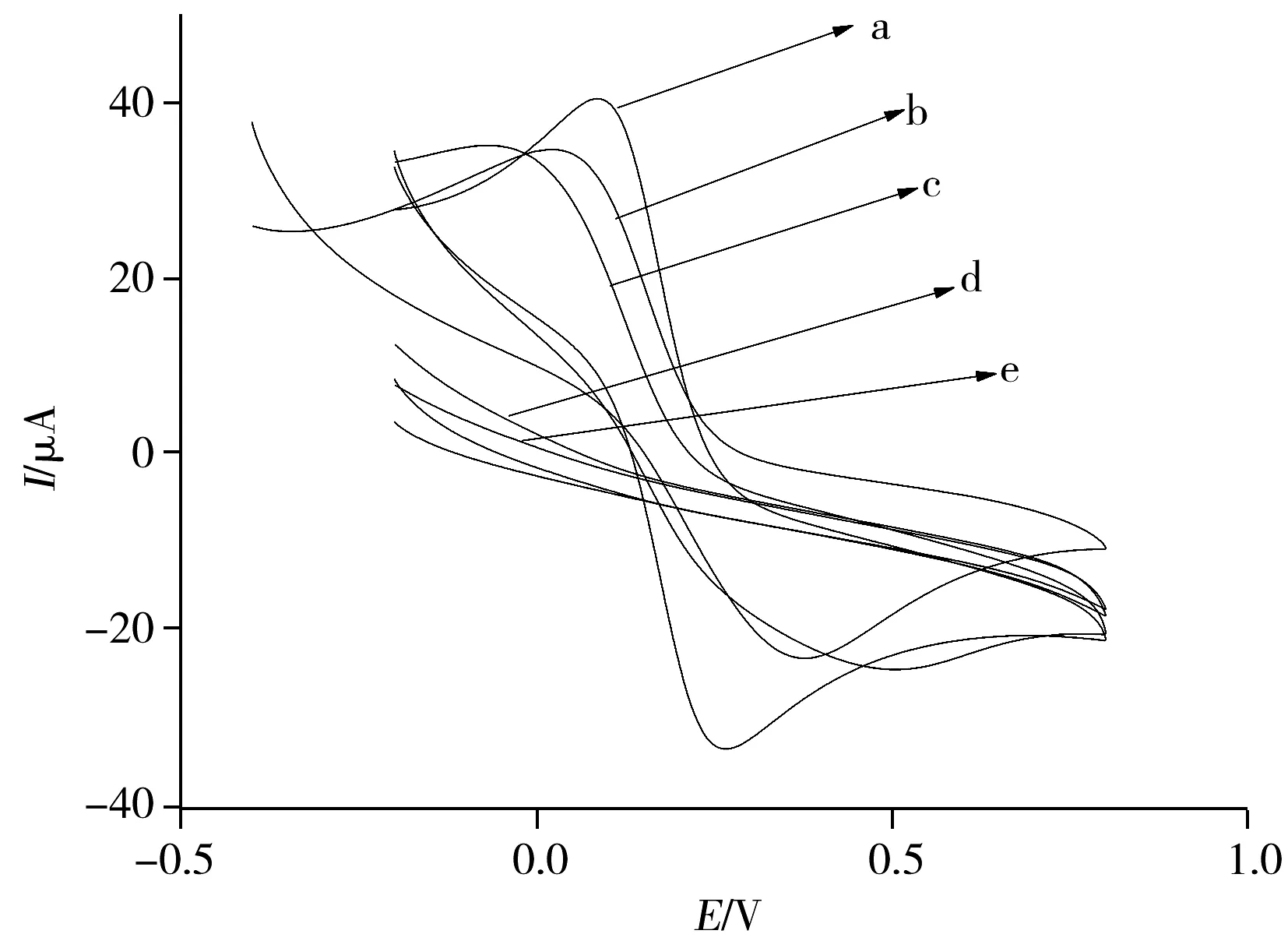
圖2 不同類型電極在鐵氰化鉀探針溶液中的CV圖Fig.2 CV curves of different electrodes in potassium ferrohydride probe solution a.GO/GCE;b,c.MIP/GO/GCE and NIP/GO/GCE after template removal,respectively;d,e.MIP/GO/GCE and NIP/GO/GCE before template removal,respectively
為了研究SA是否洗脫完全,考察了石墨烯修飾玻碳電極(GO/GCE)、洗脫前后的MIP/GO/GCE和洗脫前后的NIP/GO/GCE在K3[Fe(CN)6]探針溶液中的循環伏安圖(如圖2)。結果顯示,GO/GCE的峰電流響應值最大(曲線a),未洗脫的電極MIP/GO/GCE(曲線d)和NIP/GO/GCE(曲線e)幾乎無氧化還原峰,說明此電極基本絕緣,這是因為未洗脫的電極不會留下“空穴”,無法發生電子傳遞。洗脫后的電極MIP/GO/GCE,其峰電流響應顯著增大(曲線b),這是因為印跡分子經洗脫劑洗脫后,在印跡膜上形成與模板分子匹配的“空穴”,形成電子傳遞的通道,因而導致峰電流信號明顯加強。而洗脫后的MIP/GO/GCE表面覆蓋膜雖具有“空穴”,但較GO/GCE的傳遞性能差,所以其電流響應的強度小于GO/GCE,但大于洗脫后的NIP/GO/GCE(曲線c)[17]。
2.3 不同類型印跡電極的掃描電鏡圖
采用掃描電鏡分別對GO/GCE(A)、未洗脫的MIP/GO/GCE(B)、洗脫后的MIP/GO/GCE(C)和吸附SA平衡的MIP/GO/GCE(D)進行表征,結果如圖3所示。從圖A可以看出石墨烯的鱗片狀結構;由圖B可觀察到聚合膜已將石墨烯的鱗片狀結構覆蓋,其表面比未聚合前平整;洗脫后的電極表面可觀察到大量“空穴”(圖C);吸附SA達到平衡后的MIP/GO/GCE電極,可觀察到“空穴”數量變少,表面平整(圖D),這是因為SA在靜電力作用下與活性“空穴”結合[18]。此結論與“2.2”相互印證,說明石墨烯修飾后的玻碳電極的電流響應大于其他電極,未洗脫的電極表面則覆蓋了一層不能導電的聚合物薄膜。
2.4 電化學聚合條件的選擇
2.4.1 聚合液pH值的優化在濃度為0.2 mol/L的PBS緩沖溶液中,固定SA∶o-PPD∶Py的摩爾比為1∶4∶4,用方波伏安法研究了不同pH值(6.2、6.4、6.6、6.8、7.0)對聚合的影響。結果顯示,在考察的pH值范圍內,峰電流表現為先增大后減小的趨勢,且在pH 6.6時峰電流響應值最大。說明在此pH值下,功能單體o-PPD和Py能較好的在修飾電極表面發生氧化聚合反應,其原因可能是o-PPD和Py更適宜在弱酸性環境下發生電化學聚合。 因此實驗選擇聚合液的pH值為6.6。


2.4.2 掃描圈數的優化掃描圈數直接關系修飾電極的聚合情況,通過改變掃描圈數可制備膜厚度不同的MIP/GO/GCE。在探針溶液中進行方波伏安法掃描,掃描圈數5~25,從峰電流變化情況考察電極聚合情況。結果顯示,掃描5圈時印跡電極的峰電流響應值較小,這是由于掃描時間短,在玻碳電極表面形成的聚合膜較薄,當洗脫時能夠得到的“空穴”較少[19],使得探針溶液中的[Fe(CN)6]3-能夠結合的位點少,從而導致峰電流響應值較小。隨著掃描圈數的增加,MIP/GO/GCE在探針溶液中的峰電流也增加,在掃描15圈時峰電流達到最大。繼續增加掃描圈數,MIP/GO/GCE的峰電流呈遞減趨勢。這是由于掃描時間過長時,聚合膜變厚,使得在洗脫時洗脫液不易破壞模板分子與功能單體間的氫鍵[20],從而導致洗脫效果不好,峰電流下降。因此選擇掃描圈數15為制備MIP/GO/GCE的最佳圈數。
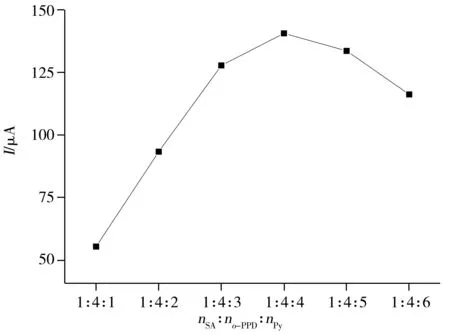
圖4 模板分子與功能單體的摩爾比對聚合的影響Fig.4 Influence of different ratios of template molecule to function monomers on polymerization
2.4.3 鄰苯二胺與吡咯的比例優化聚合液中的模板分子與功能單體的比例會影響聚合膜的結構和電化學性能,因此需考察聚合液中模板分子與功能單體的比例,以尋找最佳配比。固定SA與o-PPD的最佳摩爾比為1∶4,改變Py的用量,使得聚合液中SA∶o-PPD∶Py的摩爾比分別為1∶4∶1、1∶4∶2、1∶4∶3、1∶4∶4、1∶4∶5、1∶4∶6,考察其峰電流響應情況,結果如圖4。當SA∶o-PPD∶Py比例為1∶4∶4時,MIP/GO/GCE峰電流的響應值最大,說明此比例下有效結合位點最多,形成的印跡膜最穩定。因此,實驗選擇SA∶o-PPD∶Py的最佳摩爾比為1∶4∶4。
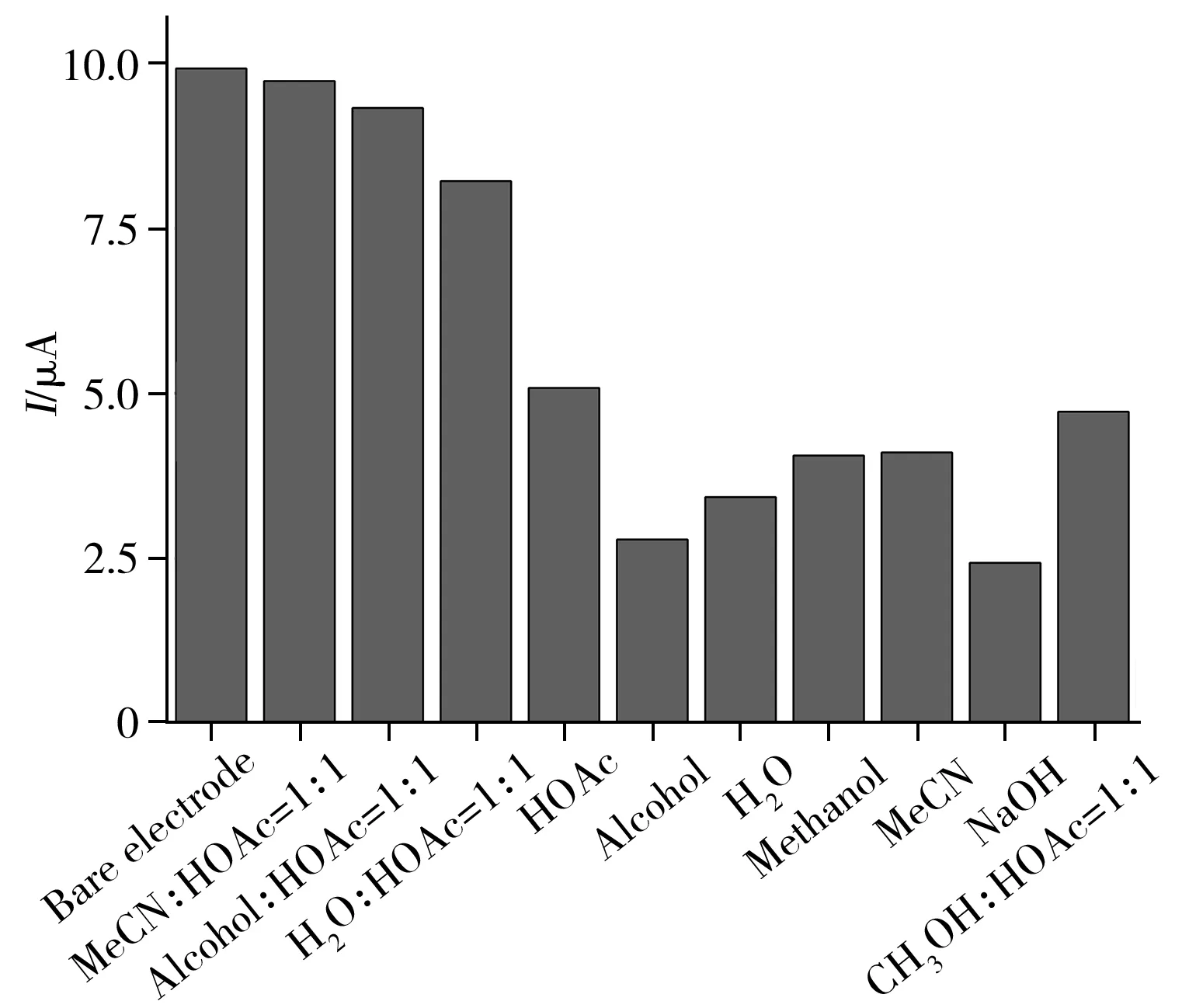
圖5 不同洗脫液種類的洗脫效果Fig.5 Effect of different types of eluents
2.4.4 洗脫液的選擇考察了混合洗脫液與單一洗脫液的洗脫效果,結果如圖5所示,采用乙腈-乙酸(1∶1,體積比)、乙醇-乙酸(1∶1)混合液洗脫SA后,MIP/GO/GCE的峰電流與裸電極的峰電流幾乎無差別,說明洗脫較完全,出現此現象可能是由于乙腈和乙醇均為優良的有機溶劑,在乙酸配合下可將大部分模板分子洗脫下來[21-22],甚至會導致印跡效果被破壞。采用用水-乙酸混合液(1∶1)雖效果略差,但相對較為溫和,不會對印跡膜造成破壞。單一洗脫液氫氧化鈉、乙醇、甲醇、水及乙酸洗脫后,峰電流響應較小(見圖5),說明洗脫不完全,效果不佳。而甲醇-乙酸混合溶液(1∶1)的峰電流響應明顯小于水-乙酸(1∶1)混合溶液,綜上,本實驗選擇水-乙酸(1∶1)混合液為最佳洗脫液。
2.4.5 洗脫時間的優化采用方波伏安法考察了不同洗脫時間對MIP/GO/GCE峰電流的影響。結果顯示,峰電流隨著洗脫時間的增加而增加,在20 min時達到平衡,故選擇最佳洗脫時間為20 min。
2.5 SA分子印跡傳感器的性能研究
2.5.1 吸附平衡時間的確定將MIP/GO/GCE放入1.0×10-6mol/L SA溶液,采用方波伏安法研究了該電極對SA的峰電流以及與吸附時間的關系。結果顯示,隨著吸附時間的增加,SA分子在MIP/GO/GCE上的峰電流響應值呈現遞增趨勢,當吸附21 min后,峰電流趨于平穩,表明吸附21 min時反應達到平衡。為了吸附更加充分,實驗選擇吸附時間為24 min。
2.5.2 線性范圍與檢出限以MIP/GO/GCE為工作電極,在濃度分別為1.0×10-8、1.0×10-7、1.0×10-6、1.0×10-5、1.0×10-4、1.0×10-3、1.0×10-2mol/L 的SA溶液中,用方波伏安法測定SA濃度與峰電流的關系。結果顯示,在1.0×10-8~1.0×10-2mol/L濃度范圍內,MIP/GO/GCE的峰電流(Y)與SA濃度的負對數(lgc)呈線性關系,線性方程為Y=6.087 5×10-6+9.201 1×10-6lgc(r=0.998 1),檢出限(S/N=3)為8.6×10-9mol/L。
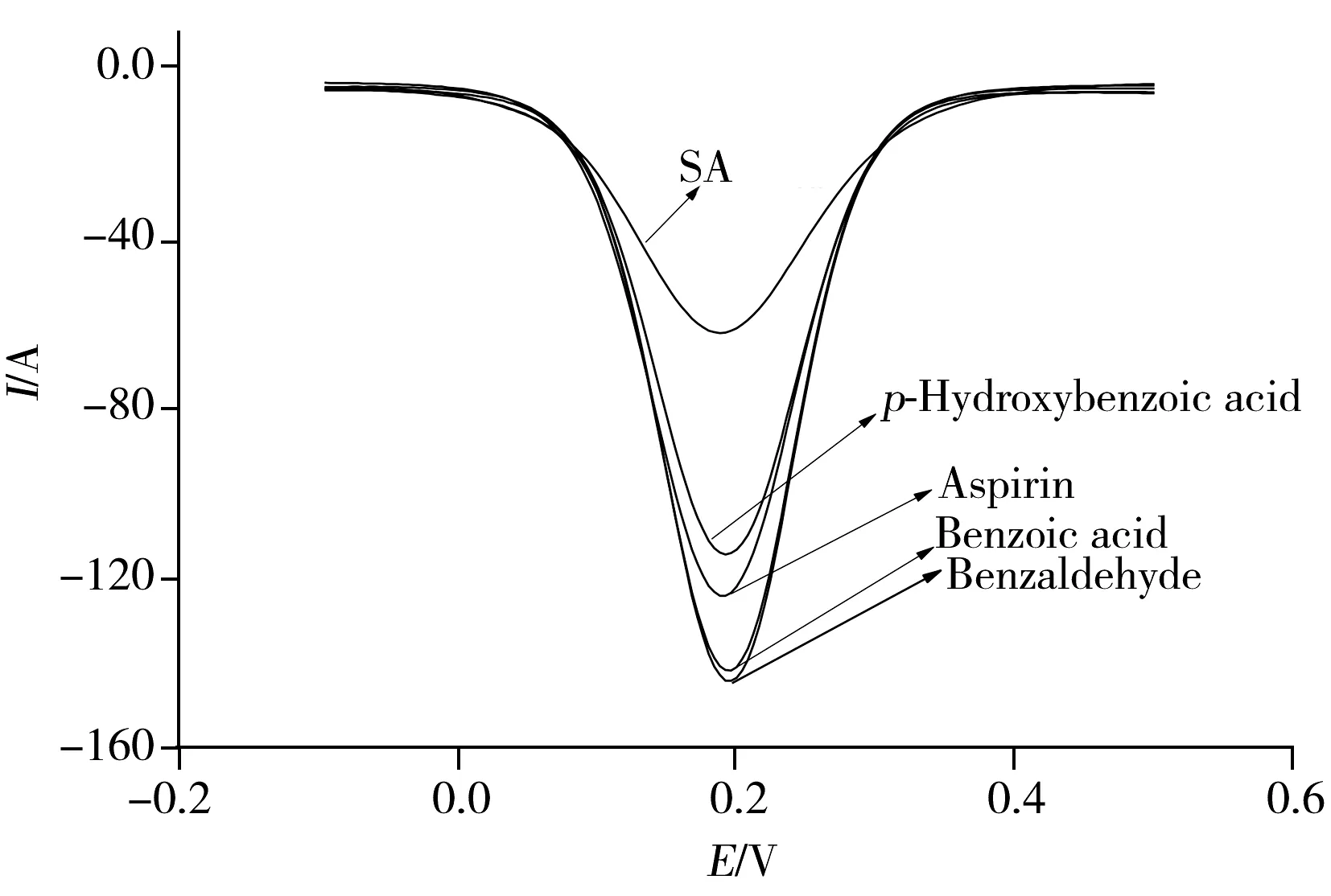
圖6 SA及其結構類似物在印跡傳感器上的響應Fig.6 Selective responses of the imprinted electrochemical sensor for SA and its structural similarities
2.5.3 方法的選擇性選擇與SA結構類似的化合物(乙酰水楊酸、苯甲酸、苯甲醛、對羥基苯甲酸)作為干擾物進行選擇性實驗。分別配制濃度為5.0×10-6mol/L的SA、乙酰水楊酸、苯甲酸、苯甲醛、對羥基苯甲酸溶液,將MIP/GO/GCE在上述溶液中浸泡吸附24 min,自然干燥后采用方波伏安法在探針溶液中掃描。結果如圖6所示,SA的峰電流響應值最小,這是因為MIP/GO/GCE上的“空穴”被SA“填滿”,能結合探針溶液中[Fe(CN)6]3-的活性位點非常少,故峰電流響應值小。取10 μL上述干擾物質溶液加入空白PBS溶液中,用方波伏安法掃描印跡電極。由于SA在PBS溶液中以陰離子形式存在,在通電時與正性的“空穴”結合形成導電性“網格”,因此其電流響應值最大,而干擾分子則因空間構型與“空穴”不匹配[23],所以峰電流響應值較小。上述研究說明所制備的MIP /GO/GCE僅對SA有較高的選擇性。
2.6 方法的重現性與穩定性
將MIP/GO/GCE置于濃度為1.0×10-7mol/L的SA溶液中,檢測其峰電流響應值,每個標準樣品平行測定6次,測得其相對標準偏差(RSD)為3.2%,說明MIP/GO/GCE的重現性較好。將MIP/GO/GCE置于pH 6.6的PBS溶液中存放1周后再檢測1.0×10-7mol/L的SA溶液,其峰電流響應值基本穩定在原來的93%左右,說明MIP/GO/GCE的穩定性較好。
2.7 混合樣品的檢測
在39 mL空白PBS溶液中加入10 mL 1.0×10-6mol/L對羥基苯甲酸干擾溶液,再加入1 mL已知濃度為1.0×10-6mol/L的SA溶液,平行測定MIP/GO/GCE 3次,測得其加標回收率分別為101%、106%、102%,相對標準偏差為3.8%,說明此傳感器可以用于實際樣品的檢測。
3 結 論
本文以SA為模板分子,o-PPD與Py作為復合功能單體,在石墨烯修飾的玻碳電極上制備對SA具有特定識別功能的電化學傳感器。研究顯示,SA在1.0×10-8~1.0×10-2mol/L濃度范圍內,分子印跡傳感器的峰電流與SA濃度的負對數呈良好的呈線性關系,其線性方程為Y=6.087 5×10-6+9.201 1×10-6lgc(r=0.998 1),檢出限為8.6×10-9mol/L。將所制備的傳感器用于混合樣品檢測,其回收率為101%~106%,相對標準偏差為3.8%。該方法操作簡便,靈敏度高,為SA的提取、分離和檢測提供了新思路。
