基于CRISPR/Cas9 技術快速構建PRV gE 基因缺失病毒
2020-05-09 01:04:34
中國動物檢疫 2020年5期
(內蒙古農業大學獸醫學院,農業農村部動物疾病臨床診療技術重點實驗室,內蒙古呼和浩特 010018)
偽狂犬病病毒(pseudorabies virus,PRV)屬于皰疹病毒科,會導致成年母豬流產、產死胎以及仔豬中樞神經系統疾病,未免疫母豬產下的仔豬感染PRV 時,死亡率可達100%[1-2]。早在1947 年,我國首次從患貓體內檢測出PRV。1970 年后,PRV 在我國呈蔓延趨勢,直至從匈牙利引入Bartha-K61 株疫苗后,疫情才得到有效控制[3]。2011 年以來,PRV 在我國發生變異,使Bartha-K61 疫苗不能提供足夠的保護,導致偽狂犬病迅速流行,并席卷全國[4-7],其中母豬患病率達35%以上,野毒感染豬群超過50%,對我國養豬業造成了巨大損失[8]。PRV 基因組編碼許多蛋白,其中gE 蛋白是主要的毒力因子。在PRV 變異株感染的豬群中,檢測發現gE基因抗體水平逐年上升。因此,gE基因缺失疫苗可以在降低病毒毒力的同時區分野毒感染和疫苗免疫[9]。我國最初引入的Bartha-K61 株就是gE基因缺失疫苗株。早在2000年,何啟蓋等[10]使用TK-/gG-/LacZ+突變株進行動物實驗,證實PRV 基因缺失毒株的安全范圍比親本毒株廣,具有很好的免疫原性。自2013 年以來,CRISPR/Cas9 技術作為強大的基因編輯技術[11],已經在病毒基因編輯上獲得了廣泛應用,如:Van等[12]利用CRISPR/Cas9 技術成功編輯了HCMV和HSV-1;2018 年穆艷霞等[13]使用CRISPR/Cas9技術,利用先重組EGFP 標簽反向篩選的方法成功獲取了IBR-ΔgE病毒;湯艷東等[14]構建了螢光素酶標記基因、EGFP 基因雙標簽重組病毒,同時建立了PRV 大片段缺失平臺,證實了2 個sgRNA介導的大片段基因缺失效率更高。因此,本研究基于CRISPR/Cas9 技術與實驗室分離到的PRV 毒株,快速對其gE基因進行編輯,旨在為今后的PRV 基因編輯提供思路,也為后期應對PRV 變異株的基礎研究及防控提供一種候選方法。
1 材料與方法
1.1 材料
1.1.1 病毒與細胞系 PRV-1 毒株,由內蒙古農業大學獸醫學院傳染病教研室分離、鑒定及保存;PK-15(豬腎細胞)、VERO(非洲綠猴腎細胞),由內蒙古農業大學傳染病教研室保存。
1.1.2 主要試劑及載體 胎牛血清(fetal bovine serum,FBS),DMEM 培養基,opti-MEM 以及EDTA-胰酶,購自Gibco 公司;BbsI 限制性核酸內切酶,購自NEB 公司;PrimeSTAR GXL DNA Polymerase 高保真酶、solution I,購自TaKaRa 公司;DNA 提取試劑盒、膠回收試劑盒,購自Therom公司;病毒基因組DNA/RNA 提取試劑盒,購自TIANGEN 公司;低熔點瓊脂糖,購自Sigma 公司;轉染試劑Lipofactamine 3 000,購自Thermo Fisher公司;大腸桿菌DH5ɑ 感受態細胞、Stbl 3 化學感受態細胞,購自北京全式金生物技術有限公司;pcDNA3.1載體、pSpCas9-2A-puro 和pSpCas9(BB)-2A-GFP 載體,由內蒙古農業大學傳染病教研室保存。
1.2 方法
1.2.1 PRVgE基因克隆測序 根據NCBI 上已公布的偽狂犬病毒(PRV)基因序列(登錄號KU056477.1),在primer premier 5.0 軟件上設計針對gE基因的上下游引物PRV-gE-F、PRV-gE-R(表1);以本實驗室提取的PRV-1 基因組為模板,采用PCR 方法擴增gE基因。PCR 反應體系:ddH2O 15 μL、GXL buffer 5 μL、dNTP 2 μL 以及PRV-gE-F、PRV-gE-R 各1 μL,PRV-1 基因組0.75 μL,GXL DNA Polymerase 酶0.25 μL。PCR反應條件:94 ℃預變性120 s;98 ℃變性10 s,60 ℃退火15 s,68 ℃延伸120 s,35 個循環;68 ℃終延伸10 min,4 ℃保存。PCR 產物送華大基因有限公司測序。
1.2.2 野生病毒感染力測定 將PRV-1 病毒按照10-1~10-8進行10 倍倍比稀釋后,各吸取100 μL 病毒稀釋液,接種于長到80%左右的PK-15 細胞上,每組8 個重復,同時設置陰性對照。逐日觀察并記錄結果,5 d 后按照Reed-Muench 法進行TCID50測定。計算公式為:
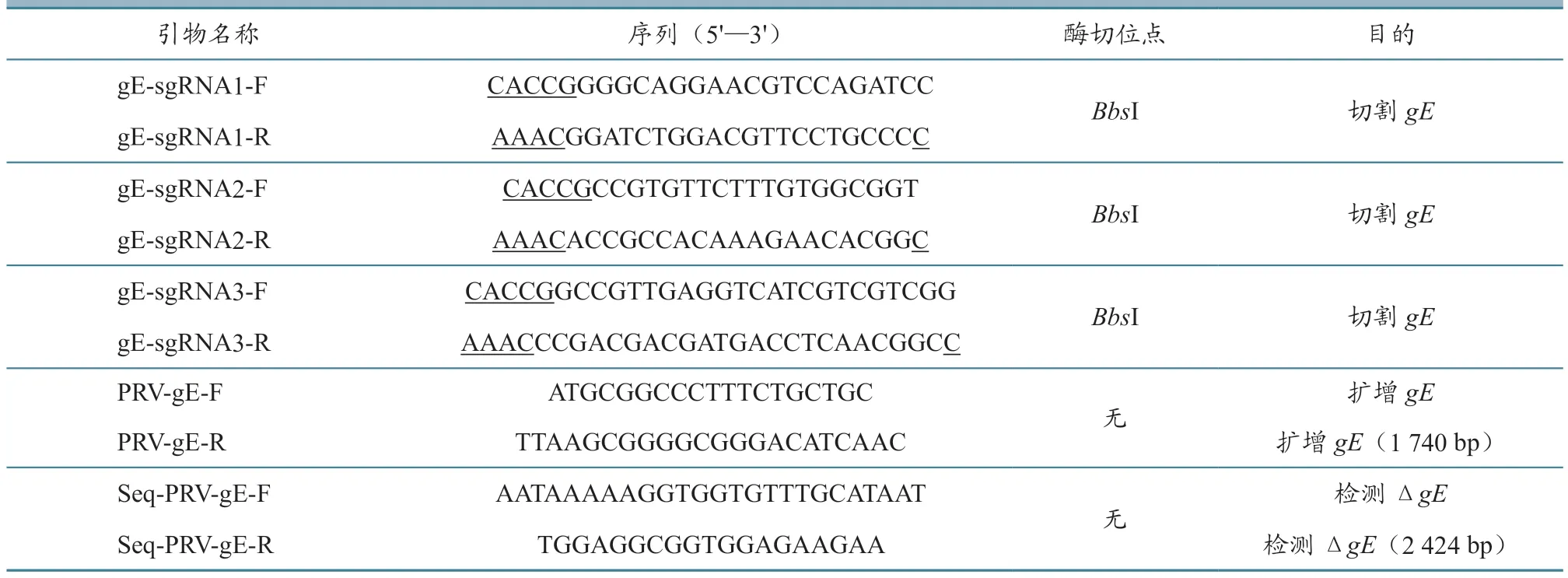
表1 試驗所用引物及目的

將比距加在高于50%死亡稀釋度的對數上,即為0.1 mL 所含病毒液的稀釋度。
1.2.3 轉染細胞系篩選 首先將pSpCas9(BB)-2A-GFP 載體,利用脂質體轉染法(Lipofectamine?3 000),將PX459-1、PX459-2 和PX459-3 質粒分別轉染PK-15 細胞與VERO 細胞,48 h 后熒光顯微鏡(ZEISS、HAL100)下觀察,篩選轉染效率較高的細胞系。
1.2.4 CRISPR/Cas9 載體構建及篩選 按照1.2.1測序得到PRVgE基因序列,通過網站http://crispr.mit.edu/設計3 對分數較高且GC 含量約50%的gE-sgRNA,并合成其互補引物對(表1);將互補的gE-sgRNA 引物gE-sgRNA1-F/gE-sgRNA1-R、gE-sgRNA2-F/gE-sgRNA2-R 和gE-sgRNA3-F/gEsgRNA3-R 進行95 ℃ 5 min 高溫變性,然后逐漸降溫至16 ℃,獲得3 條雙鏈sgRNA;以BbsI 雙酶切pSpCas9-2A-puro 載體,膠回收大的載體片段;將3 條雙鏈gE-sgRNA 連入pSpCas9-2A-puro 酶切回收的載體中,使用Solution I 16 ℃過夜連接,將連接產物轉化Stbl 3 感受態細胞,提取質粒,對測序結果使用snapgene 軟件分析,獲得CRISPR/Cas9 重組質粒PX459-1、PX459-2 和PX459-3。將構建好的PX459-1、PX459-2 和PX459-3 分別轉染VERO 細胞,12 h 后接種MOI=0.1 的PRV-1,感染48 h 后使用10%中性甲醛固定2 h,之后使用0.8%結晶紫染色0.5 h,沖洗干凈后觀察。
1.2.5 PRV-1-ΔgE構建 利用脂質體轉染法,將CRISPR/Cas9 載體PX459-1、PX459-2,按 照1:1 的比例轉染入VERO 細胞培養6 h,更換含2%FBS 的細胞維持液,培養至12 h 后接種MOI=0.1的PRV-1,待細胞病變至90%左右時收毒;將病毒反復凍融3 次,按照1:1 000 稀釋度稀釋感染6 孔板中的PK-15 細胞,病毒吸附2 h 后,使用DMEM(含2% FBS、1%低熔點瓊脂糖)覆蓋,置于37 ℃、5%的CO2培養箱中培養48 h;于超凈工作臺中使用槍頭隨機挑起10 個病毒蝕斑,分別置于200 μL DMEM 中,反復凍融3 次后接種生長至90%的PK-15 細胞。重復以上步驟,噬斑克隆純化5 代,然后將病毒接種于PK-15 細胞中擴大培養,提取病毒基因組,使用檢測引物seq-PRVgE-F/R 進行PCR 擴增檢測。PCR 體系與反應條件與1.2.1 中介紹相同。將測序結果使用MEGA 7 軟件進行對比分析,鑒定后擴大培養,-80 ℃保存。
1.2.6 PRV-1-ΔgE遺傳穩定性檢測 將PRV-1-ΔgE接種至PK-15 細胞,連續傳代至第20 代,并取第5、10、20 代缺失病毒DNA,使用PRV-gE-F和PRV-gE-R 進行PCR 擴增。PCR 產物送華大基因測序,并對測序結果進行比對。
2 結果與分析
2.1 PRV-1 gE 基因PCR 擴增
按照1.2.1 中PCR 體系獲得的gE基因測序結果為1 740 bp,與試驗預期一致(圖1)。
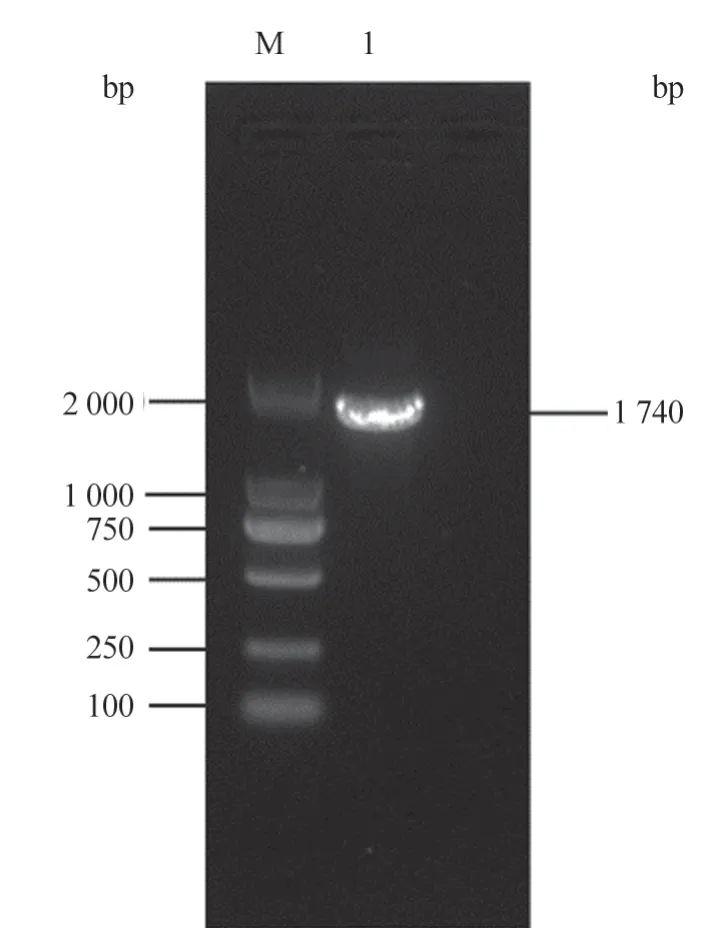
圖1 PRV gE 基因PCR 擴增結果
2.2 PRV 野毒感染力測定
野生病毒感染力測定如表2 所示。當病毒稀釋至10-6時,出現CPE 的細胞孔所占百分比高于50%,為76.9%,稀釋至10-7時,出現CPE 的細胞孔所占百分比低于50%,為36.4%。
將表2 所測得數據帶入1.2.2 中的公式,得到的比距約為0.7,加上高于50%感染的稀釋度對數(10-6),PRV-1 的TCID50為10-7.7/mL。
2.3 轉染細胞系篩選
通過熒光顯微鏡觀察,發現VERO 細胞轉染效率高于PK-15 細胞(圖2),因此本試驗選擇VERO 細胞作為轉染細胞系。
2.4 CRISPR/Cas9 質粒構建與篩選
根據snapgene 軟件分析結果,判定CRISPR/Cas9 質粒PX459-1、X459-2 和PX459-3 構建成功(圖3)。將PX459-1、X459-2 和PX459-3 質 粒轉染細胞篩選,發現sgRNA1 和sgRNA2 效率大于sgRNA3(圖4),因此選擇sgRNA1 和sgRNA2做后續試驗。
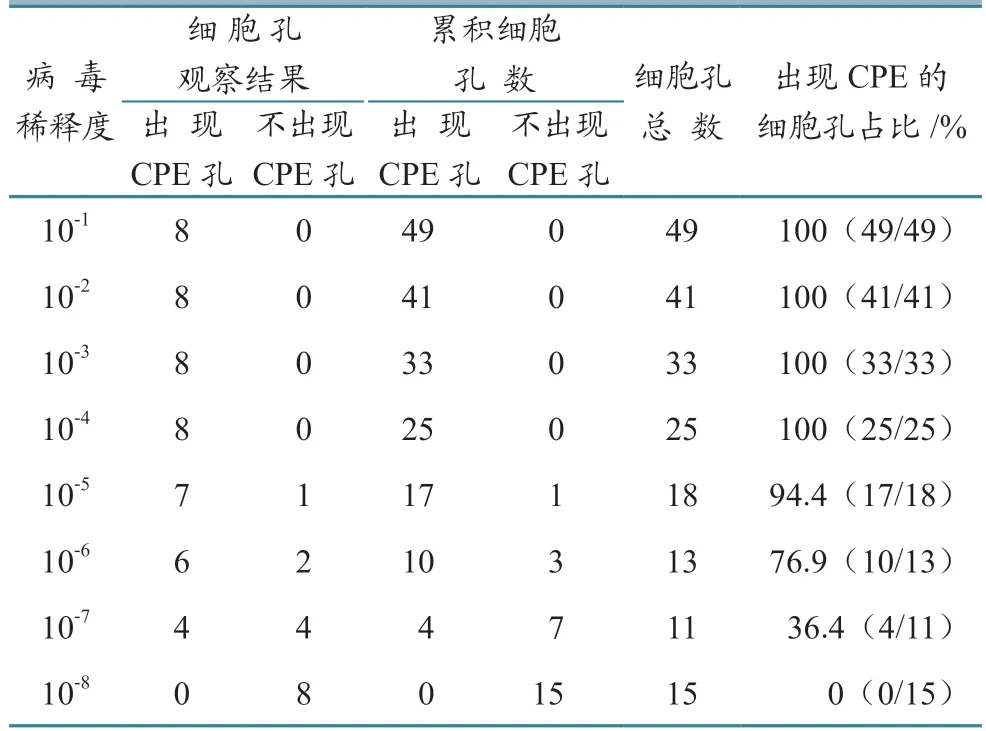
表2 病毒TCID50 測定結果

圖2 pSpCas9(BB)-2A-GFP 在PK-15 與VERO 細胞上的轉染效率(50×)
2.5 PRV-1-ΔgE 構建
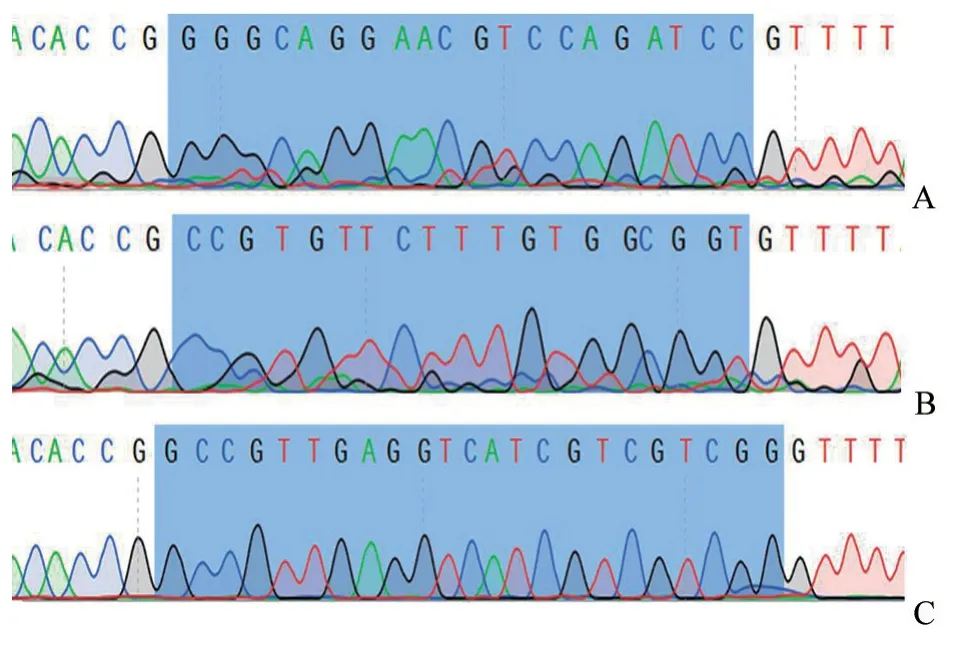
圖3 sgRNA 構建測序結果

圖4 sgRNA 篩選結果
使用seq-PRV-gE-F/R 引物進行PCR 檢測,發現gE基因發生缺失(圖5);將測序結果使用MEGA 7 軟件比對分析發現,PCR 產物大小為2 424 bp,與預期一致,PRV-1-ΔgE為2 132 bp,gE基因323~613 bp 位置缺失291 bp(圖6)。
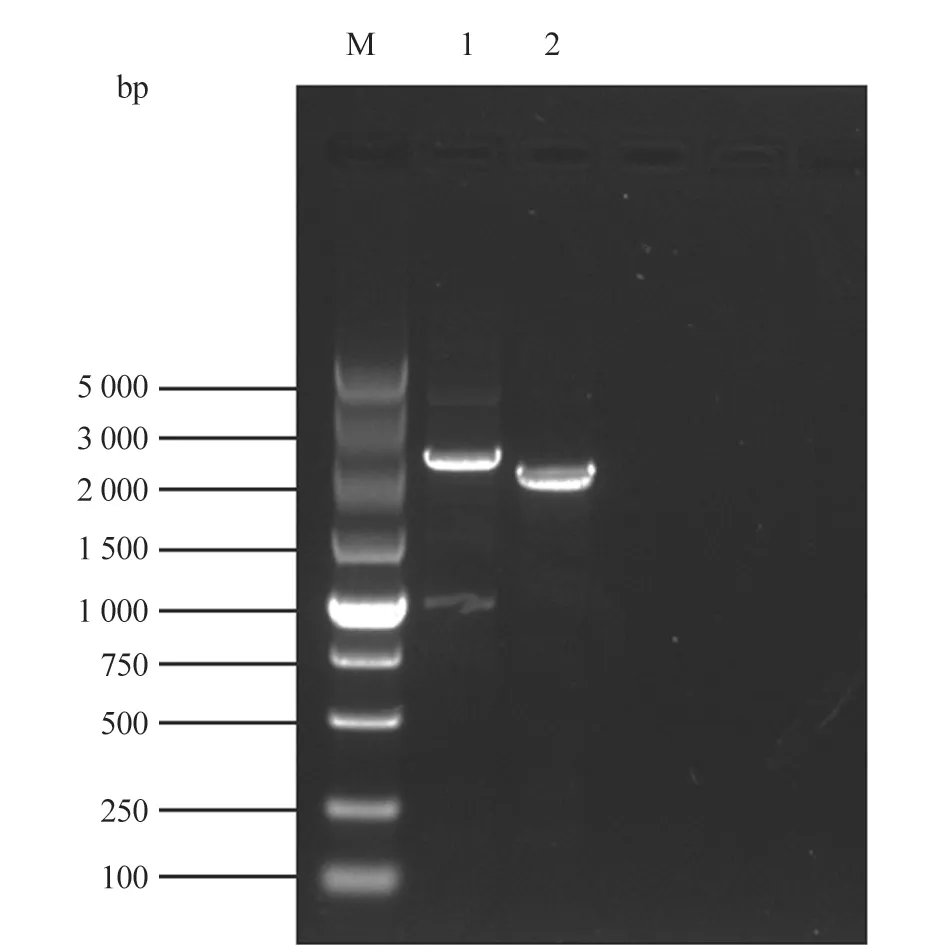
圖5 PRV gE 基因檢測引物PCR 鑒定結果

圖6 PRV-1 與PRV-1-ΔgE 測序比對結果
2.6 PRV-1-ΔgE 遺傳穩定性檢測
將PRV-1-ΔgE連續傳代至20 代,分別取5、10、20 代病毒進行測序分析,發現沒有發生堿基突變現象。
3 討論
目前,針對PRV 感染還沒有系統有效的治療方法,因此疫苗接種是控制其流行和凈化的有效方法。自2011 年以來,PRV 不斷發生變異,至今依然在我國部分地區廣泛流行[15-16],因此快速應對新發變異的PRV 就顯得尤為重要。這些基因的缺失會導致PRV 毒力降低,且不影響病毒的感染和復制能力。據相關資料[17-18]顯示,一些歐美國家通過基因缺失疫苗免疫,已經根除了PRV。PRV 基因工程疫苗研制通常選用缺失TK、gE、gI或gG等毒力基因[19-20]。
穆艷霞等[13]使用CRISPR/Cas9 技術編輯IBRV,在gE基因位置處重組EGFP 熒光標記,通過sgRNA 反向敲除EGFP 基因構建IBR-ΔgE病毒。吳鳳筍[21]使用傳統的同源重組結合Cre/loxp系統,先在gE、TK位置分別重組EGFP 標記基因,篩選攜帶熒光的PRV,后將標記基因分別切除構建了1 株PRV-ΔgE-ΔTK重組病毒。靶向基因編輯技術與傳統的同源重組技術相比較,它對基因組的修飾效率可以提高103~105倍[22]。因此,本研究基于CRISPR/Cas9 技術,僅通過單獨轉染1 對sgRNA,接種病毒誘導細胞內非同源末端修復即獲得PRV-1-ΔgE。相比較于傳統的同源重組技術,CRISPR/Cas9 技術大大提高了基因編輯篩選速度與效率。首先,在CRISPR/Cas9 技術當中,篩選轉染效率高的細胞系與獲得高效的sgRNA至關重要。因此,本試驗首先選擇了PK-15 細胞與VREO 細胞作為轉染細胞系,經過篩選發現VERO 細胞的轉染效率遠高于PK-15 細胞且可以被PRV 感染。本試驗結果與湯艷東[13]、穆艷霞[14]等細胞系篩選結果一致。其次,sgRNA 的工作效率及脫靶效率直接影響基因編輯的成功率。Cho 等[23]認為成對的Cas9 內切酶共同作用于目標基因組,會使脫靶效率明顯降低。在sgRNA 篩選中,本試驗首先設計了3 條sgRNA,經過噬斑形成試驗,篩選到2條可以高效切割病毒基因的sgRNA,最后在噬斑克隆過程中,因為沒有標簽,所以隨機篩選了10個單獨噬斑克隆。在這個過程中,要挑選相對于野生病毒噬斑稍小的噬斑克隆,突變毒株的噬斑會稍小于野生病毒[14]。同時,相比較于標簽反向篩選,本試驗沒有插入標簽,因而后期就省去了刪除標簽的步驟。
4 結論
綜上,本試驗采用CRISPR/Cas9 第3 代基因編輯技術,快速對PRV-1 進行編輯,確定了1對高效編輯PRVgE基因的sgRNA,并獲得1 株PRV-1-ΔgE。本研究提供了一種高效的PRV-1 毒株編輯方法,為后續構建多基因缺失病毒提供了基礎數據,也為快速應對PRV 變異及其相關變異株基礎研究提供了新思路。
