藥物中元素雜質檢測技術研究最新進展
2020-05-09 09:09:10姚尚辰許明哲尹利輝胡昌勤
分析測試學報 2020年4期
朱 俐,趙 瑜,姚尚辰,許明哲,尹利輝,胡昌勤
(中國食品藥品檢定研究院,北京 102629)
藥物中的元素雜質是藥品質量控制和保證(QC/QA)的重要組成部分,也是一個關注熱點[1-6],根據藥品監管機構和藥典規定,測定原料藥、輔料、藥物制劑及生產過程中每個階段的元素雜質并將其控制在可接受的限度范圍內是非常重要的[7]。元素雜質的測定不僅可以快速明確地表征藥物生產環節的污染情況,而且可以規范藥品質量,保證藥品安全[8]。藥物活性成分(API)、原輔料及中間體都可能含有金屬、非金屬或類金屬元素。藥物中的元素雜質可能是非故意引入,比如As、Cd、Hg、Ni、Pb元素可通過原料(水、試劑和合成的API)或輔料(穩定劑、填充劑、粘合劑、顏料等)引入[9-15];還有可能是非故意性帶入,如Cr、Cu、Mo、Ni、V,可能來源于藥物與反應容器或制藥設備(混合罐、過濾器和灌裝線等)的表面相互作用,此外包裝和儲存過程中元素雜質的浸出也可能無意引入元素雜質[16]。在API合成中加入重金屬催化劑也可導致藥物制劑中存在痕量或超痕量的元素雜質,如Ir,Os、Pd、Pt、Rh和Ru元素[17]。
藥物制劑中的元素雜質由于存在潛在毒性和可能引起的健康風險而備受企業和消費者的關注[18]。藥物中的元素雜質對患者沒有任何治療作用,并可以催化API降解,從而縮短藥物的保質期,產生副作用或不良反應,損害人體健康。因此,藥品生產過程中使用的各種原輔料、水以及反應中間體都需要監控元素雜質,這樣才能確保藥品元素雜質水平符合規定,保證藥品的安全性和有效性[19]。美國藥典(USP)和國際協調委員會(International Conference on Harmonization,ICH)對元素雜質都有了新的分析和控制策略[20]。與早期的重金屬分析方法相比,新的分析策略不僅擴大了檢測元素范圍而且降低了檢出限。目前我國藥品元素雜質檢測方法依然采用具有百年歷史的比色法,其原理為金屬離子與硫化物離子反應顯色,再與標準鉛溶液(質量濃度為1.0×10-3mg/L)進行目視比色[21]。該方法缺乏特異性、靈敏性和準確性,存在主觀目測顏色誤差;并且只能得到樣品中元素雜質的總量值,不能對單個元素雜質進行定量分析。ICH Q3D元素雜質指南提出了元素雜質的分類和控制要求。隨著中國加入ICH,意味著中國也要遵循ICH指南和標準。ICH Q3D提出對藥物中的元素雜質需進行定性和定量分析評估,并限制最低元素雜質含量。元素雜質已越來越受到制藥企業和藥品監管機構的關注,為了適應新的法規和標準,現代化的儀器分析技術已經逐步代替老舊的方法。本文總結了近年來藥物中元素雜質的分析方法,以期為藥品中元素雜質的質量分析提供新的思路。
1 藥品質量控制與不同分析技術的應用
藥品質量控制指整個生產過程中的質量評估,包括原料、API和藥品、包裝材料等。藥物不同的物理化學性質、基質的多樣性及新法規的限量標準,對分析方法提出了更高的要求,即雜質分析需達到μg/g和ng/g水平,對應的理想分析方法的檢出限應低于ng/g。如此低的檢出限不僅要有更靈敏的分析儀器,而且對儀器選擇性也提出了更高的要求,因為在較低的濃度范圍內可能存在很多雜質。各國藥典均有收載的比色法屬于定性分析,已經不能滿足目前元素雜質分析的要求,必然會被高靈敏度的分析方法所替代,如電感耦合等離子體發射光譜(ICP-OES)和電感耦合等離子體質譜(ICP-MS),不僅分析速度快、檢出限低,且可以同時分析多種元素,工作曲線范圍寬[22-24]。目前原子吸收光譜法(AAS)、X射線熒光光譜法(XRF)、儀器中子活化分析(INAA)、ICP-OES和ICP-MS等方法已經用于藥物中金屬雜質的分析。ICP-OES和ICP-MS是ICH Q3D推薦的元素雜質檢測方法。基于等離子的分析技術,如微波等離子體發射光譜(MP-OES)、ICP-飛行時間質譜儀(ICP-TOF MS)、高分辨等離子體質譜(HR-ICP-MS)和同步ICP-MS具有更強大的功能,也已用于藥物的元素分析[25-28]。還有更新的技術,如激光誘導擊穿光譜(LIBS)和激光燒蝕ICP-MS(LA-ICP-MS)也開始在制藥行業的元素分析中得到更廣泛的應用。
1.1 用于快速篩查的便攜式分析儀器
在藥品研發、生產、質量控制及銷售的每個階段,都需要進行快速元素篩查。快速篩查方法最好不需要樣品制備或者只需進行簡單的樣品制備,分析速度要快,且檢出限能達到藥品標準的要求,最重要的是儀器便攜。早期由于缺少便攜式分析儀器,快速篩查方法發展緩慢,但近幾年來已經研制出很多便攜式、手持式光譜儀,采用此類光譜儀進行藥品快速篩查可取得較好的效果。目前,用于藥品生產過程和終端產品定性篩查的有4類便攜式儀器:便攜式X熒光光譜儀(XRF)、離子遷移譜(IMS)、拉曼光譜和近紅外光譜(NIR),并分別用于檢測重金屬、保健食品中的化學藥品和藥物成分中的有機污染物。NIR光譜還可以用來定性和定量鑒別假藥和非法仿制藥[29],LIBS也可以替代便攜式XRF用于重金屬快速篩查[30-31]。雖然便攜式XRF的檢測限較高且無法檢測原子序數小于12的元素,但仍廣泛作為快速篩查技術用于制藥行業。Arzhantsev等[32]建立了基于連續小波變換濾波器的XRF分析新方法,并將該方法用于手持式XRF光譜儀測定藥物材料中的有毒金屬,無需在測量前測量校準樣品。尹利輝等[33]采用便攜式XRF建立了膠囊中鉻的快速篩查方法,不僅通量高而且可以實時檢測,適用于藥品生產過程控制和上市藥品的質量監督。
1.2 常規測試方法
目前中國藥典仍采用傳統的比色法進行重金屬(Ag、As、Bi、Cd、Cu、Hg、Mo、Pb、Sb和Sn)檢測,該方法通過有色重金屬硫化物沉淀來確定金屬雜質的含量:首先將藥品在500~600 ℃(800 ℃,歐洲藥典)熾灼使之灰化,調節pH值后加入硫代乙酰胺顯色,最后通過目視與經同樣方法處理的0.001%(10 μg/ mL)鉛標準溶液進行顏色比較。盡管該方法在制藥工業中被廣泛接受和使用,但由于基于硫化物沉淀顯色的方法是非特異性的,其靈敏度低,耗時耗力,且回收率很低或根本沒有回收率[9],這類傳統過時的方法必將被先進的儀器分析方法所取代。
1.3 紫外光譜法
紫外-可見(UV-Vis)分光光度法由于相對經濟、快速和簡便而被廣泛采用。對于固定光程,UV-Vis分光光度法可用于測定本身對紫外可見光有吸收的金屬元素。采用UV-Vis測定時,可用配體選擇性地結合金屬離子(如鐵(Ⅱ)和銅(Ⅱ))形成有更高摩爾吸光系數的有色絡合物,從而提高在不同藥物中的檢測靈敏度[34]。Janwadkar等[35]以2′,4′-二羥基苯乙酮縮氨基脲作為萃取劑,用分光光度法測定了藥物樣品中的Ce(Ⅵ),該方法不僅可以測定各種合金、礦石,還可以測定藥物中的特定有機金屬化合物。Basotra等[36]開發出一種新型分光光度法,該方法將藥物與鄰苯二胺絡合,通過檢測706 nm處綠色復合物的吸光度,用于鹽酸順鉑片的精確測定,方法準確性可達99.98%。
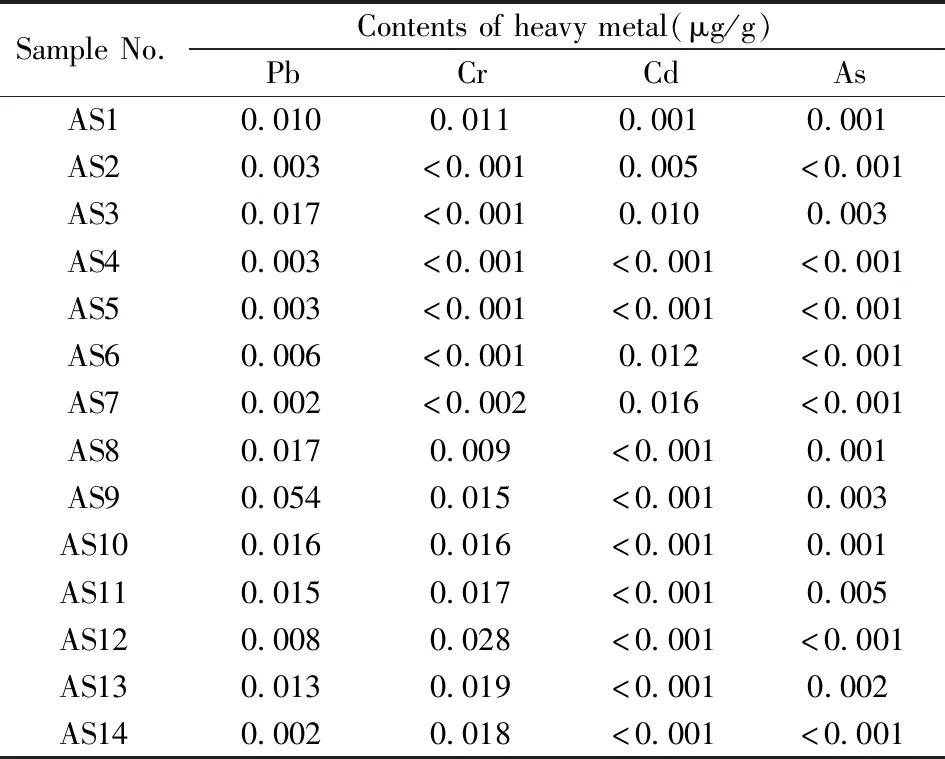
表1 市售阿司匹林制劑中有毒重金屬雜質含量的AAS測定結果[45]Table 1 Determination results of toxic heavy metal impurities in commercially available aspirin preparations by AAS[45]
1.4 原子吸收光譜法(AAS)
常用的火焰原子吸收光譜法(F-AAS)可用于測定不同材料包括藥品中μg/mL及以下含量的金屬元素[37-38]。石墨爐原子吸收(GF-AAS)由于樣品全部參與原子化,且避免了原子濃度在火焰氣體中的稀釋,分析靈敏度得到了顯著提高,適用于分析超痕量(ng/mL)的金屬元素并能用于少量樣品的分析和固體樣品的直接分析。汞可以采用冷原子吸收法檢測,而一些類似As、Sb的“揮發性”元素可以通過氫化物法進行檢測[39]。冷原子吸收尤其適用于檢測痕量汞,通過測定254 nm處的吸收檢測藥物中的汞,檢出限可達9 ng/mL[40],冷原子吸收的主要優點是可以將汞從基質中完全分離[41]。F-AAS和GF-AAS均可在藥物質量控制中可靠地檢測金屬雜質。Gomez等[42]通過AAS監測了貫葉連翹藥物衍生物中Ca、Cu、K、Li、Mg、Mn、Na、Ni和Zn的含量,張珉娜等[43]用火焰原子吸收光譜監測翁瀝通膠囊里的銅用以評估藥品的質量,王梅等[44]采用微波消解-石墨爐原子吸收光譜測定了布洛芬原料藥中的微量金屬雜質。表1列出了由AAS確定的各種市售阿司匹林制劑中有毒重金屬雜質的含量(μg/g)[45]。AAS技術也有其局限性,比如,F-AAS的靈敏度較低,對于痕量、超痕量分析需進行富集和分離;不能進行多元素同時分析;標準工作曲線的線性范圍窄等,但該方法由于專屬性好等優點仍然應用廣泛。
1.5 X射線熒光光譜法(XRF)
XRF由于樣品制備簡單、可多元素同時分析而逐漸受到廣泛關注。XRF是利用初級X射線光子或其他微觀離子激發待測物質中的原子,使之產生熒光(次級X射線)而進行物質成分分析和化學態研究的方法。樣品可以是固態、粉末和液態。XRF是非接觸分析,因此不會產生記憶效應,且XRF檢測無需破壞樣品,因此樣品可以回收再利用。目前,波長色散型X射線熒光光譜儀和能量色散型X射線熒光光譜儀均已被應用于藥物API中Zn、Fe和Ni的測定[46]。Marguí等[47]采用波長色散型X熒光光譜儀檢測藥物中的金屬雜質(Fe、Zn、Cr和Ni),并根據歐洲藥品評價機構(EMEA)和ICH的規范對其進行了驗證。Kikongia等[48]采用波長色散型XRF,通過擬合回歸的方法測定了藥物原料中的痕量重金屬Ni、Cr、Pb ,并取得了良好的結果。Arnet等[49]采用XRF測定了8種頭孢曲松仿制藥中的元素雜質,在8種仿制藥中都檢出了Br和Zn,而在原研藥中未檢出元素雜質。Linder等[50]使用二芐基二硫代氨基甲酸酯(Dibenzyldithiocarbaminate)作為共沉淀劑,用XRF測定了藥物中常見的12種金屬元素。全反射XRF也可以測定藥物中的痕量元素,Wagner等[51]采用全反射XRF研究了卵磷脂、胰島素和不同來源的普魯卡因和色氨酸中的金屬元素,并根據元素濃度的微小差異區分了不同批次、不同生產工藝或純化過程的藥物。相較于其他傳統的分析儀器,XRF具有較高的檢出限,因此在藥廠中應用較少,也不適用于對藥物中元素雜質的準確定量。
1.6 儀器中子活化分析法(Instrumental neutron activation analysis,INAA)
INAA是一種相對簡單的分析技術,可用于測定各種材料中的元素豐度。該方法通過鑒別和測試樣品因輻照感生的放射性核素的特征輻射,進行元素和核素分析,以γ射線分光儀測定光譜,根據波峰分析確定樣品成分后,基于輻射能的強弱進行定量分析。最近50年,該技術在環境、地質、植物、食品和藥物的微量元素分析工作中得到了廣泛應用,其優點有:(1)樣品無需處理可直接測試;(2)所需樣品量非常少,通常只需幾毫克;(3)很多元素的檢出限在ng/g范圍內;(4)可同時檢測超過40種元素。由于上述優點,在電ICP-MS出現之前,INAA深受分析工作者的喜愛。Chen等[52]早在1993年就用INAA測定了天然藥物樣品中的14種微量元素(K、Sc、Cr、Fe、Co、Zn、Br、Rb、Cs、La、Sm、Eu、Au和Th)。Choudhury等[53]采用中子活化分析技術分析了中草藥中的31種元素,Kamath等[54]使用ICP-AES和INAA技術測定了一些傳統印度藥物中的汞。放射性藥物在注冊前需對有毒重金屬元素進行定量,Hernndez[55]使用INAA精確地測定了3種放射性藥物(六甲基丙叉二胺肟(HMPAO),二巰基丁二酸(DMSA)和二乙三胺五乙酸(DTPA))中的40種微量和痕量元素。盡管INAA具有很多優點,但因為采用該法測定時需要反應堆,存在放射性,且需要對不同半衰期的核素進行分次測量,使得大部分元素的分析周期較長,從而限制了其廣泛使用。
1.7 電感耦合等離子體發射光譜法(ICP-OES)
ICP-OES是利用高頻電感耦合產生等離子體放電的光源進行原子發射光譜分析的方法,通過將樣品的發射光譜強度與已知濃度的標準強度進行比較,獲得未知樣品中的相應元素濃度[56-57]。該法最大的優點是可在很寬的線性范圍內同時高靈敏地檢測約60種元素[58-59]并且具有較寬的動態線性范圍。美國藥典新通則USP<233>推薦ICP-OES和ICP-MS為元素雜質的檢測方法。Stoving等[20]應用ICP-OES法測定了藥片中As、Cd、Cu、Cr、Fe、Hg、Ir、Mn、Mo、Ni、Os、Pb、Pd、Pt、Rh、Ru、V和Zn的含量。楊永健等[60]以格列齊特為研究對象,采用分組的方式,克服了元素之間的光譜干擾,建立了化學原料藥中19種金屬雜質的ICP-OES檢測方法。采用ICP-OES分析元素含量時,由于分析譜線的重疊,在測量過程中可能會發生光譜干擾而影響檢測結果,為了排除干擾,需更換無干擾的分析譜線。然而,有時不存在完全沒有潛在光譜干擾的波長,如Ir、Os、Pb、Pt和Rh。此外,記憶效應可能導致誤差很大,如 Os易被氧化成OsO4并在樣品引入系統中積累。類似有記憶效應的另一個元素是Hg,因此要注意控制進樣量,避免引入高濃度的汞而導致結果誤差。最后,由于制備的樣品溶液中易于電離的元素(EIE)的含量高,如Ca、K、Mg和Na,可能導致出現化學干擾,特別是對于含有不同礦物賦形劑的藥物或腸胃外溶液[61]。
1.8 電感耦合等離子體質譜法(ICP-MS)
相較于ICP-OES,ICP-MS更適用于痕量和超痕量元素分析,檢出限可達10-12g/mL。ICP-MS線性范圍寬,譜線簡單,分析速度快速,靈敏度高,可以定量和半定量,幾乎可以分析所有元素[62],并且該方法所需的樣品量非常少,樣品溶液濃度較低,從而最大程度地減少了可能出現的基質效應。自1980年首次將ICP離子源與四極桿質譜連接之后,其商用儀器也于1983年問世。應用四極桿質譜進行元素分析會相應降低分析能力,主要是由于藥品中的基質元素會產生多元素干擾,如樣品中存在C、Cl、S和P元素時會導致等離子體中形成多原子離子,干擾As、Cr、Cu元素的測定[63]。隨著ICP-MS技術的快速發展,碰撞/反應池和離子阱分析器技術可完全消除或減少多原子光譜干擾。因此,ICP-MS已經成為元素痕量分析的重要手段。Wang等[9]采用藥典中規定的重金屬比色法和ICP-MS分析了藥物中的痕量元素,并指出了重金屬比色法的局限性,建議采用ICP-MS替代過時的“重金屬限度檢查”。Lásztity等[7]采用ICP-MS測定馬來酸依那普利中的Pd、亞葉酸鈣中的Pt和左旋多巴中的Rh,檢測限分別為15、2.8、2.5 ng/g。Murty等[5]通過ICP-MS測定了雙環胺、乙胺丁醇、吡嗪酰胺和呋喃唑酮類藥物中的重金屬含量,通過選擇合適的同位素,分析藥物中的Ti、Cr、Mn、Fe、Co、Ni、Cu、Zn、Cd、Hg、Pb含量,發現Cr、Fe、Ti和Cu分別在雙環胺、乙胺丁醇、吡嗪酰胺和呋喃唑酮中含量最高,4種藥物中均不含Ni和Hg,而乙胺丁醇和吡嗪酰胺中存在痕量的Cd。Pinheiro等[64]采用ICP-MS測定了9種藥品中ICH Q3D規定的23種金屬元素(除Os外),回收率為72%~128%,9種藥品的元素雜質含量均符合USP<232>的規定。許雯雯等[65]采用ICP-MS法測定了鹽酸安舒法辛緩釋片中7個元素雜質的含量,其相關系數均大于0.99,加標回收率為92.2%~104%,重復性RSD≤1.2%(n=6),精密度RSD≤3.0%,滿足USP<233>方法學驗證的要求。樣品的檢測結果遠小于ICH Q3D中各元素雜質的每日允許暴露量(PDE)。張磊等[66]用ICP-MS測定了氟哌酸藥物中的21種元素雜質,汞在0~10 ng/mL范圍內線性關系良好,其他20種目標元素在0~100 ng/mL范圍內線性關系良好,r均大于0.999 0,平均回收率為80%~120%。Lewen等[67]建立了藥物中金屬元素的ICP-MS快速篩查方法,可對藥物中的As、Se、Cd、In、Sn、Sb、Pb、Bi、Ag、Pd、Pt、Hg、Mo和Ru進行篩查,回收率在89%~102%之間。Balaram[68]采用硝酸消解樣品,通過ICP-MS測定了4種市售藥物中34種元素雜質的濃度,結果表明ICP-MS可作為藥物中元素雜質的監測方法。
2 樣品制備
根據樣品類型,USP<233>提出的藥物元素分析可以采用ICP-OES和ICP-MS兩種方法[69]進行,這兩種方法不僅可以提高檢測能力和靈敏度,而且減少了樣品制備步驟。由于ICP-OES和ICP-MS需要將樣品消解,因此在元素雜質分析之前的樣品制備是關鍵步驟。通常情況下,元素雜質在復雜的基質中以低濃度存在,因此在樣品制備中應防止元素雜質的損失及二次污染。
USP<233>規定了樣品制備的4種主要優選方法(圖1),這些樣品制備方法適用于ICP-OES和ICP-MS,可降低樣品污染或目標元素損失的風險。但是,由于藥物樣品的多樣性,USP<233>未給出詳細的步驟,因此樣品制備具體方法的選擇取決于樣品的性質[69]。值得注意的是,在選擇合適的溶劑溶解或消解樣品時,需考慮樣品的化學穩定性和揮發性,還應考慮樣品中目標元素的穩定性,避免目標元素的損失,比如汞元素和鉑族元素。因此USP<233>規定,對這些元素進行分析檢測時,需在樣品溶液中加入適當的穩定劑,以保證其穩定性[69]。
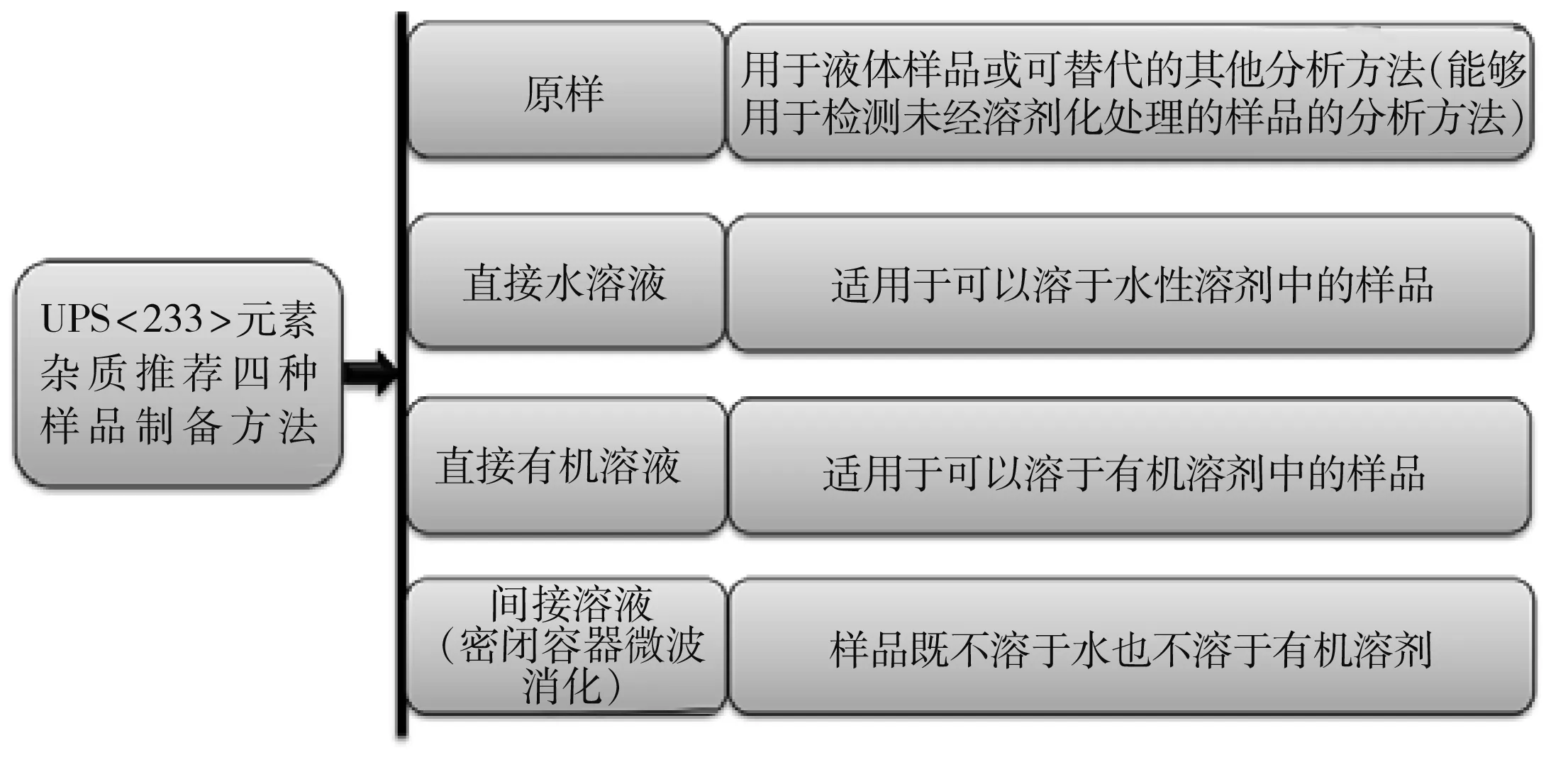
圖1 USP<233>中元素雜質測定時樣品的制備方法Fig.1 Preparing method for detection elemental impurity according to USP<233>
2.1 直接溶解法
ICP-OES和ICP-MS的樣品制備優先選用無機酸或者有機溶劑水溶液簡單溶解的制備方法[63,70-72]。根據USP<233>,可溶性固體和液體樣品可用稀硝酸(HNO3)溶解,如Lásztity等[7]采用0.2 mol/L HNO3處理樣品,通過ICP-MS測定了馬來酸依那普利中的鈀、亞葉酸鈣中的鉑和左旋多巴中的銠;Wang等[9]采用1% HNO3溶解樣品,建立了簡便、快速、靈敏的原料藥、中間體和藥物中元素雜質的定量-半定量方法,Antes等[63]用5%HNO3稀釋場外營養液,采用ICP-MS法測定了其中的元素雜質。也有研究將HNO3和HCl混合處理樣品,如Van Hoecke等[71]用0.009 mmol/L KBrO3(含有1%(體積分數)HNO3和1%(體積分數)HCl)微波輔助消解法測定了藥物輔料中的元素雜質。Fischer等[70]以1%HNO3和0.15% HCl水溶液消解樣品后用ICP-MS快速分析了藥品中的元素雜質。
消解樣品時加入0.1%~1%HCl可以絡合Hg2+[70-71]、Pd2+[4,71]、Os2+[71]和Pt4+[11],確保這些離子在溶液中穩定,避免延遲效應和記憶效應。Au3+也可以起到與HCl類似的作用,穩定Hg2+和降低記憶效應[19]。也可以直接采用濃硝酸溶解來制備樣品,如經7 mol/L HNO3[7]、80%HNO3[4,9,17]消解之后可以直接檢測[9]或者用水稀釋降低酸的濃度后檢測[7]。除了液體進樣,ICP-OES/MS還可以采用懸浮液霧化進樣[73],Zachariadis等用0.5 mol/L的稀硝酸溶解樣品,加入0.5%聚乙二醇辛基苯基醚(Triton X-100)表面活性劑作為穩定劑和分散劑,使樣品成為漿液后直接進樣,對粉末狀抗生素中的多元素雜質進行了分析檢測[11]。Dennis等[16]采用鹽酸-氯化鉀緩沖液、磷酸鹽緩沖液,在pH 3.0和pH 9.0條件下,以甲醇-水體系提取藥品包裝材料,痕量檢測了藥包材中的32種元素雜質。
對于不溶于水的樣品,可用有機溶劑進行溶解,如含有2%乙二胺四乙酸鈉(EDTA)的二甲基甲酰胺(DMF)[4]、2-丁氧基乙醇∶水(1∶3,體積比)[8]、二乙二醇單乙醚(DGME)(加入1%硫代乙酰胺作為Pb絡合劑)[72]、乙醇[74],經溶解、超聲后,不需要進一步處理樣品即可直接檢測。
直接溶解樣品的方法簡單、操作步驟少,減少了待測元素的損失,對于快速篩查和常規監測元素雜質非常重要。采用ICP-OES分析樣品需考慮總固體溶解量(TDS),常規分析中,分析溶液的TDS越低越好,一般控制在1%左右,TDS太高易導致炬管堵塞,Lásztity[7]、Lewen[8]、Wang[9]、Jia[17]、Fischer[70]、Al-Ammar[72]等采用了0.1%的樣品濃度,Antes等[63]采用了0.4%和0.5%的樣品濃度。較低的樣品濃度不會造成儀器過載,同時也降低了基體效應,減少了譜線干擾。也有文獻報道采用較高的TDS,比如1%[71]、2.5%[11,63,72]的樣品濃度,這與檢測使用的儀器和檢測對象有關。
2.2 間接溶解法
因為藥品由API和一些輔料組成,如粘合劑、著色劑、顏料等,這些物質很難采用直接溶解的方法消解,因此這樣的樣品需要采用濕法消化間接溶解。濕法消化間接溶解的關鍵是要確保樣品完全溶解并且有較低的炭殘留(RCC)[75],重要的是最終樣品溶液的酸度應該與參比溶液保持一致,因為參比溶液的酸度較高時,分析信號會降低[75]。濕法消化有可能會導致揮發性元素的損失,如Hg[76],因此這類樣品最好采用微波消解法消解。相對于敞開式濕法消化,微波消解采用密閉的消解罐,避免了樣品在消解過程中揮發性組分的損失,也避免了外部環境的污染,大多數藥品都可以只用HNO3消解[77]或者HNO3和H2O2混合消解。Raghuram等[10]用HNO3∶H2O2(7.5∶1,體積比)消解樣品,采用ICP-OES分析了藥物活性成分中的23種金屬元素;Zachariadis[11]用5∶1的HNO3和H2O2消解,ICP-OES法分析了抗生素中的21種元素雜質,但值得注意的是,H2O2可能會因為試劑的純度問題引起樣品污染[78-79]。也可以用HNO3/H2SO4[80],或HNO3/HCl[71,81-82],或HNO3/HCl/H2O2消解樣品[82]。當樣品中有二氧化硅(SiO2)、二氧化鈦(TiO2)或者滑石粉(Mg3Si4O10(OH)2)時,需加HF消解樣品,比如HNO3∶HCl∶HF(3∶3∶1,體積比)[15]和HNO3∶HCl∶HF(2∶2∶1,體積比)[82]。
消解之后的樣品一般用純水進行稀釋[13,70,77],但由于Hg、Os和Pd在氧化性溶劑中揮發而不穩定,可采用稀鹽酸(<0.5%)稀釋[70-71,82],且 Os和Pd因為受樣品基質的影響,稀釋時需額外加入穩定劑以保證結果的準確性,比如含有硫脲(0.01 mol/L)和抗壞血酸(0.1 g/L)的0.5%乙酸[83],含有0.009 mmol/L KBrO3[71]或者0.01 mol/L硫脲[70]的1%HCl。
無論采用哪種方法制備樣品,均需加標回收以確保方法的可行性和準確性。
3 結論與展望
元素雜質控制是藥品質量安全保證的重要研究內容之一,根據《美國藥典》的新通則USP<232>、USP<233>和ICH Q3D的元素雜質指南,現行的重金屬檢測方法在檢測范圍、準確性、靈敏度和專屬性等方面均不能滿足要求,其必將被ICH Q3D推薦的ICP-MS和ICP-OES所取代。ICP-MS似乎是最適合于藥物中元素分析的技術手段,但ICP-MS靈敏度非常高,因此存在的干擾也比ICP-OES多,實際應用中如何消除這些干擾是關鍵。相比較于ICP-MS,ICP-OES雖然靈敏度較低,但干擾相對較少,并且具有操作簡便,高通量等優點,因此選擇哪一種方法分析元素雜質還要考慮檢測目的和檢測限度的要求。
隨著中國正式加入ICH,中國藥品的監管政策也必須遵循ICH的指南和標準。2015版《中國藥典》重金屬檢查主要用于金屬總量的控制,在中國與國際接軌的新形式下,下一版中國藥典必將出現元素雜質的檢測方法。符合ICH Q3D要求,是中國創新藥走向國際市場的硬性條件,無論是中國制藥企業還是監管部門,根據ICH Q3D的不同元素PDE值和不同服藥途徑,建立或評審原輔料或藥物成品的元素限度,已經勢在必行。
猜你喜歡
中國合理用藥探索(2022年1期)2022-11-26 00:22:32
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
中國衛生(2016年5期)2016-11-12 13:25:28
海峽科技與產業(2016年3期)2016-05-17 04:32:12
中國衛生(2015年5期)2015-11-08 12:09:48
