含能功能化氧化石墨烯的制備、熱分解行為及其對AP熱分解的催化作用
2020-05-13 12:16:00沈心怡張蓉仙劉祖亮郝堯剛蘇宏平
火炸藥學報 2020年2期
成 健,沈心怡,王 睿,張蓉仙,劉祖亮,郝堯剛,蘇宏平
(1. 浙江工業大學安全科學與工程系,浙江 杭州 310014; 2. 江蘇大學化學化工學院,江蘇 鎮江 212013;3. 南京理工大學化工學院,江蘇 南京 210094; 4.甘肅銀光化學工業集團有限公司,甘肅 白銀730900)
引 言
低特征信號推進劑可顯著提高戰略、戰術導彈武器系統的生存能力和突防能力,是21世紀戰略、戰術導彈系統的首要發展目標[1]。高氯酸銨(AP)被廣泛應用于現代戰略、戰術導彈武器系統的固體火箭發動機。AP高溫分解溫度越低,固體推進劑的點火延遲時間越短,燃速越高[2-3]。研究表明,金屬、金屬氧化物、金屬復合物等惰性燃燒催化劑,以及含能燃燒催化劑對AP熱分解具有明顯的催化作用[4-5]。然而,含金屬化合物的固體推進劑在燃燒過程中將產生明顯的特征信號[5],同時排放含金屬(如Cu和Pb)的有毒有害物質,對環境造成嚴重污染。因此,含金屬型燃燒催化劑不能滿足低特征信號AP基固體推進劑未來的發展要求。
氧化石墨烯(GO)具有二維平面層狀結構,每一層GO單片上含有許多含氧官能團OH、C—O—C、C—O和COOH,因此GO具有許多優于石墨烯的性能,如良好的潤濕性和兩親性等。此外,GO單片上的含氧官能團對熱刺激敏感,易發生熱分解反應,生成rGO,并釋放大量的熱,表現出一定的含能特性。近年來,將GO作為含能添加劑、鈍感劑和燃燒催化劑載體應用于推進劑領域已有大量文獻報道[6]。研究表明,GO對AP熱分解反應表現較低的催化活性[7-10]。將GO與金屬、金屬氧化物等復合,可得到GO基納米復合物,其催化活性顯著優于GO以及相應的金屬和金屬氧化物[8]。由此可見,將GO基納米復合物作為新型燃燒催化劑具有一定的應用前景,但其仍然為金屬化合物,且能量水平不高,也不能滿足低特征信號AP基固體推進劑未來的發展要求。
通過對GO表面含氧官能團進行功能化修飾,可以拓寬GO的應用范圍并進一步提升相應新材料的性能,具有重要的研究價值[11]。由文獻的研究結果可知,GO可對AP熱分解反應表現一定的催化活性[7-10]。同時,劉子如[12]研究發現,HMX可加速AP熱分解,主要原因是HMX熱分解產物NO2可氧化AP的離解產物NH3,進而促進AP的高溫分解反應。基于以上研究結果,可推測對GO進行含能功能化修飾,在其片層表面引入含有—NO2的功能化官能團。一方面可以提高GO的能量水平,另一方面,在GO片層表面引入含—NO2功能化官能團,含—NO2功能化官能團可能會與GO產生協同效應,賦予GO較高的催化活性。因此,對GO進行含能功能化修飾,可能是獲取新型非金屬含能燃燒催化劑的有效途徑。
為了提高GO對AP熱分解反應的催化活性,本研究以2,4,6-三硝基苯胺(TNA)作為功能化官能團,對GO進行含能功能化修飾,合成一種新型含能功能化氧化石墨烯(EFGO)。采用HRTEM、Raman、FT-IR和XPS對其結構進行鑒定;用TG-DTG,DSC和TG-IR技術研究GO和EFGO熱分解行為及GO和EFGO對AP熱分解反應的催化作用,初步評價EFGO作為非金屬含能燃燒催化劑應用于AP基固體推進劑的可行性。
1 實 驗
1.1 試劑與儀器
TNA,自制[13];N,N二甲基甲酰胺(DMF)、三乙胺(TEA),均為分析純,國藥集團化學試劑有限公司;GO,純度≥98%,厚度為1.0~1.77nm,片層直徑為10~50μm,層數為1~5層,蘇州恒球石墨烯有限公司;AP,分析純,上海邁瑞爾化學技術有限公司。
JEM-3010型HRTEM,日本JEOL公司;HR Evolution型顯微共焦激光拉曼光譜儀,法國Jobin Yvon公司;MAGNA-760型傅里葉變換紅外光譜分析儀(KBr壓片),美國NICOLET公司;PHI-5702型X射線光電子能譜儀,日本ULVAC-PHI公司;DSC823e差示掃描量熱儀,瑞士METTLERTOLED公司,試樣質量為1.0~1.5mg,升溫速率為10℃/min,升溫區間 50~500℃,N2流量為30mL/min;Diamond型熱重分析儀,美國PerkinElmer公司,試樣質量為1.0~1.5mg,升溫速率10℃/min,升溫區間50~800℃,N2流量為30mL/min。與其聯用的IR光譜儀為Nicolet iS10 紅外光譜儀,美國Nicolet公司,聯用加熱管溫度為200℃,紅外光譜儀氣體池溫度為210℃。
1.2 EFGO的制備
1.2.1 合成路線
EFGO的合成路線為:
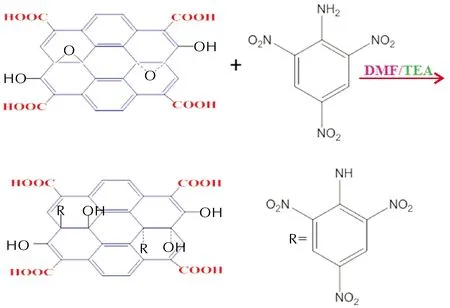
1.2.2 實驗過程
依次將0.5g GO、0.2g TNA和1mL TEA加入到40mL無水DMF中,在氮氣保護條件下將混合物加熱至100℃反應48h,反應結束后過濾,分別用甲醇和水洗滌3次,60℃真空干燥得黑色粉末0.46g。
1.3 GO/AP和EFGO/AP的制備
GO和EFGO在AP混合物中質量分數分別為2.5%、5%和10%。AP混合物的制備步驟為:分別將GO和EFGO與純AP按設定比例加入到100mL蒸發瓶中,經旋轉蒸發器常溫均速混合12h即得到本研究所需測試樣品。
2 結果與討論
2.1 EFGO的結構
C—O—C 主要分布于GO片層的表面,能夠與TNA分子中的—NH2發生開環反應,其反應機理是—NH2對α-C的親核進攻。采用HRTEM,FT-IR,Raman和XPS鑒定EFGO的結構。圖1為GO和EFGO的HRTEM圖。

圖1 GO和EFGO的HRTEM圖
由圖 1(a)和(b)可知,GO呈片狀結構,片層數較少,且厚度較薄,表面比較光滑,與文獻結果一致[12]。從圖 1(c)和(d)可以觀察到EFGO不規則的片層邊界,片層具有透明性。同時,EFGO表面出現一些褶皺,局部還發生一定程度的折疊。表明將TNA通過共價鍵修飾到GO表面,使GO的形貌發生變化,但仍然保持GO基本的框架結構。
為表征修飾前后結構的變化,分別對GO和EFGO進行Raman測試,結果如圖 2所示。
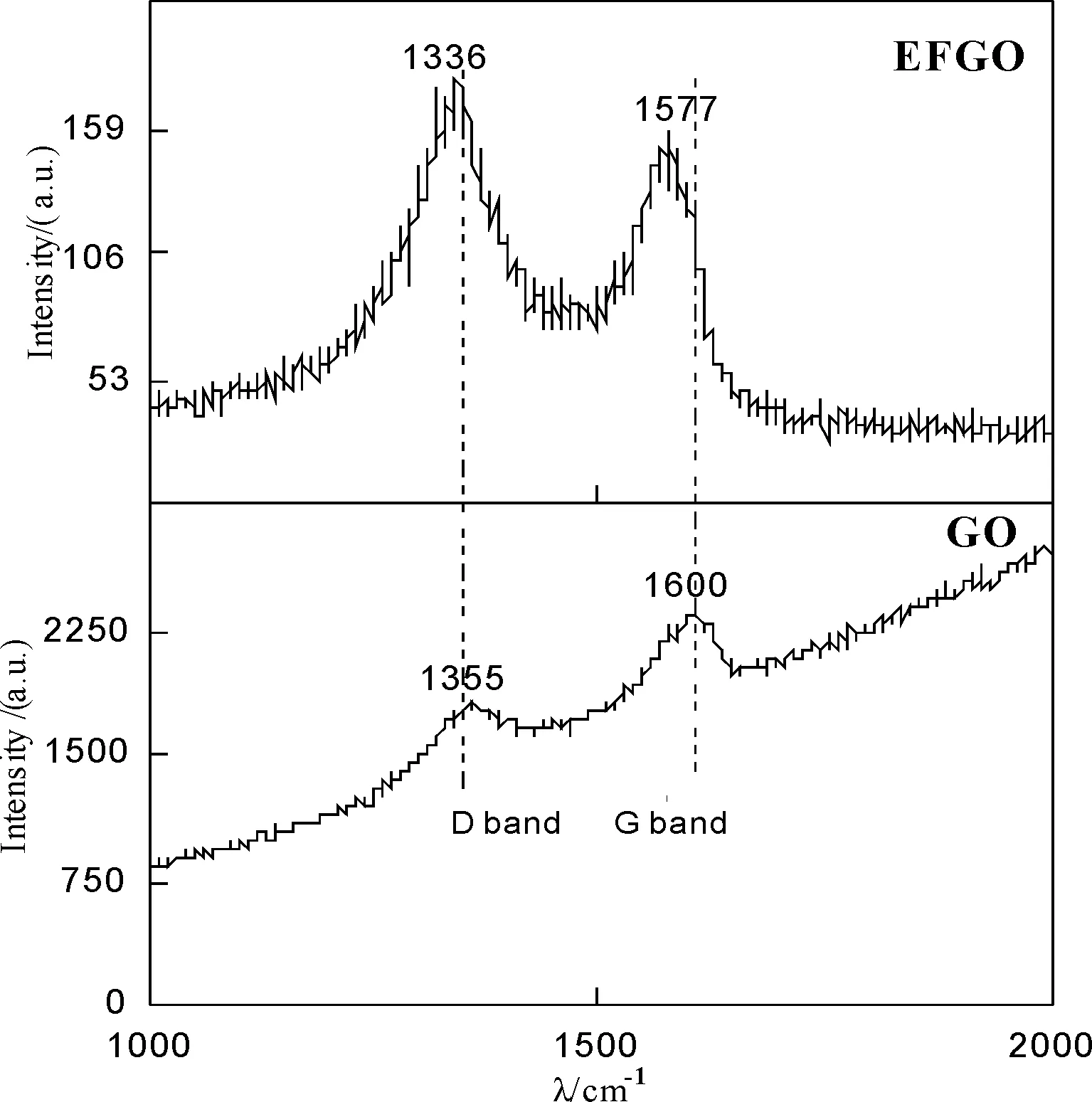
圖2 GO和EFGO的拉曼光譜圖
由圖 2可知,GO在1355和1600cm-1處顯示特征峰,分別與碳材料的D峰和G峰對應。石墨烯在1580cm-1左右的特征峰G峰是碳sp2結構所特有,反映其對稱性和結晶完整程度,位于1350cm-1左右的特征峰D峰為缺陷峰,反映石墨片層的無序性。D峰與G峰的強度比(ID/IG)可表征石墨烯共價改性程度[16]。EFGO樣品D峰和G峰的峰值相對于GO分別前移19和23cm-1,D峰與G峰的ID/IG由0.82增加至1.20,表明EFGO的框架結構相對于GO未發生明顯的改變,但片層對稱性和結晶完整程度均有一定程度的降低,無序性增大,與文獻結果一致[16]。此外,ID/IG值的變化還表明EFGO共價改性程度較高,證明GO片層表面大多數C—O—C與TNA分子中的—NH2發生共價鍵合。
為確認TNA通過共價鍵修飾至GO片層表面,采用FT-IR表征GO和 EFGO,結果如圖 3所示。
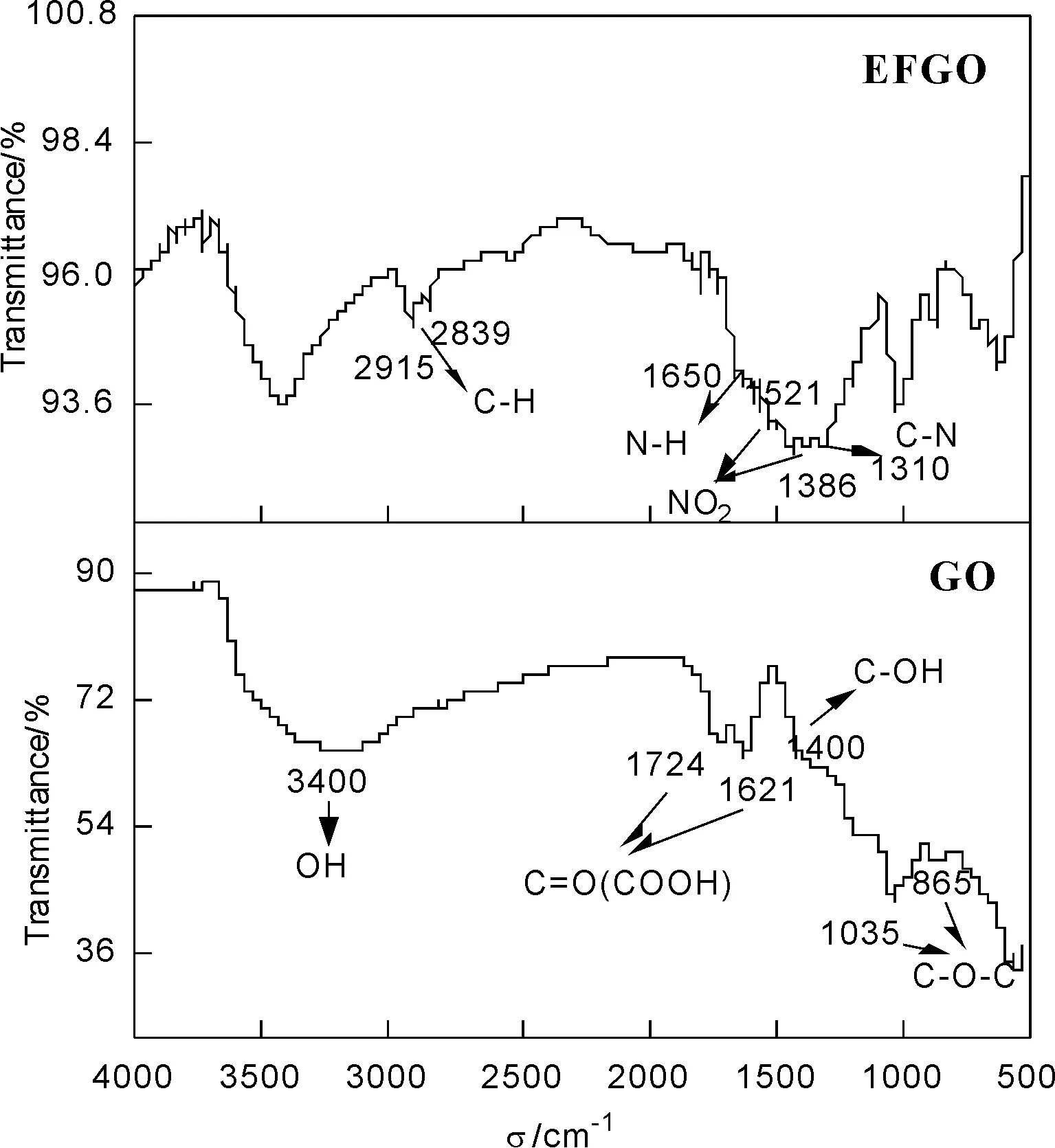
圖3 GO和EFGO的FT-IR圖譜
由圖 3可知,GO分別在3400、1724、1621、1400、1035和865cm-1處顯示吸收峰,分別對應OH伸縮振動、C—O伸縮振動、C—OH伸縮振動,OH變形和C—O—C、C—O伸縮振動。對于EFGO,在1400cm-1處C—OH吸收峰的強度增大,范圍變寬,而C—O—C對應的C—O伸縮振動峰強度減弱。此外,EFGO還分別在2900、1650、1521、1386和1310cm-1左右處顯示新的吸收峰,分別對應于TNA分子中C—H伸縮振動、N—H變形振動和—NO2的伸縮振動,以及新生成C—NH鍵的C—N伸縮振動[15]。以上變化結果證明,TNA分子中—NH2與GO片層表面的C—O—C發生開環反應,生成C—NH鍵。
為進一步鑒定EFGO的結構,分別對GO、TNA和EFGO進行XPS表征,結果如圖 4和表 1所示。
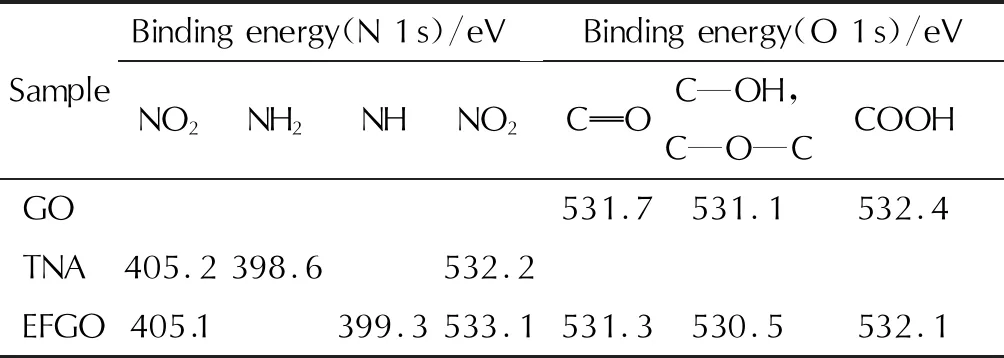
表1 GO、TNA和EFGO的XPS圖譜N 1s和O 1s分峰擬合結果
由圖 4和表 1結果可知,GO含C和O元素,O 1s分峰擬合結果表明GO分子中O元素存在4種化學狀態:531.1eV(C—OH, C—O—C)、 531.7eV (C—O)和532.4eV (COOH); TNA含C、O和N元素,N 1s分峰擬合結果表明TNA分子中N元素存在2種化學狀態:398.6eV(NH2)和405.2eV (NO2),與文獻[18]結果一致; EFGO含C、O和N元素,O 1s分峰擬合結果證明EFGO分子中的O元素存在5種化學狀態,其中4種化學狀態530.5eV(C—OH、C—O—C)、 531.3eV (C—O)和532.1eV (COOH)分別與GO分子中O元素一致。此外,O元素出現一種新的化學狀態533.1eV (NO2),與TNA分子中的—NO2一致,初步證明TNA被成功修飾至GO片層表面。從EFGO N 1s分峰擬合結果還可以觀察到,當TNA被修飾至GO片層表面后,其分子中398.6eV(NH2)擬合峰消失,由399.3eV(C—NH)代替,進一步證明TNA分子中的—NH2與GO片層表面的C—O—C發生開環反應,生成C—NH鍵。
HRTEM、FT-IR、Raman和XPS測試和分析表明,TNA通過共價鍵修飾至GO片層表面,其中,TNA分子中—NH2與GO片層表面的C—O—C發生開環反應,生成C—NH鍵。
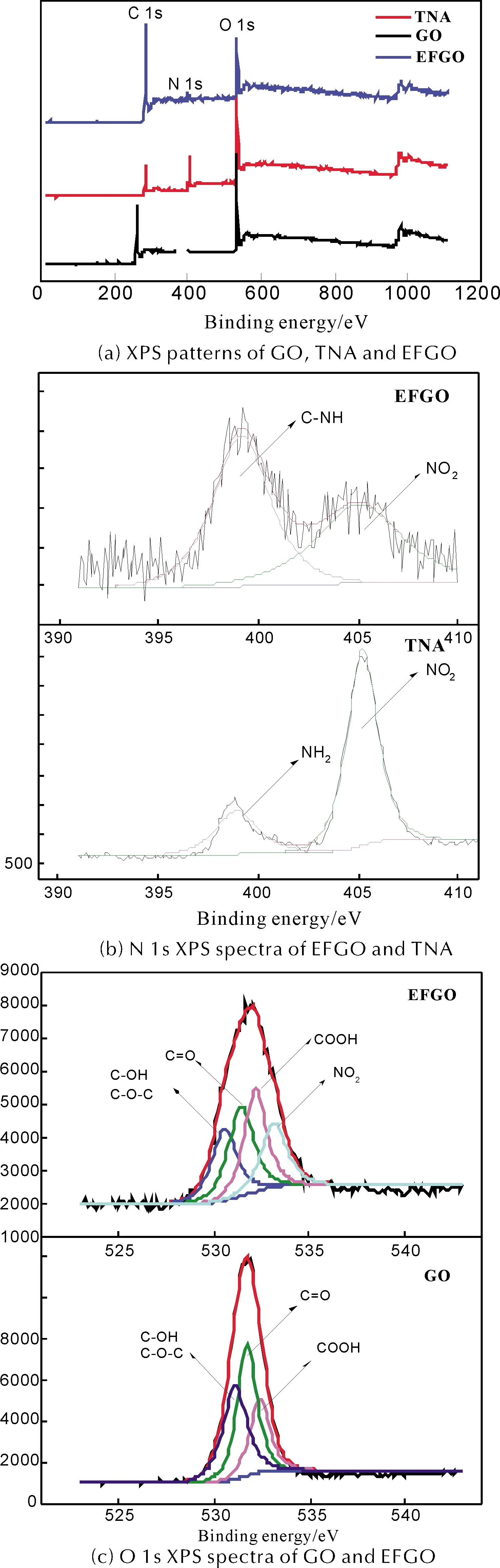
圖4 GO、TNA和EFGO的XPS圖譜以及N 1s和O 1s的分峰擬合結果
2.2 TNA、GO和EFGO的熱分解行為和熱穩定性
圖5為TNA、GO和EFGO在升溫速率10℃/min下的TG-DTG曲線。
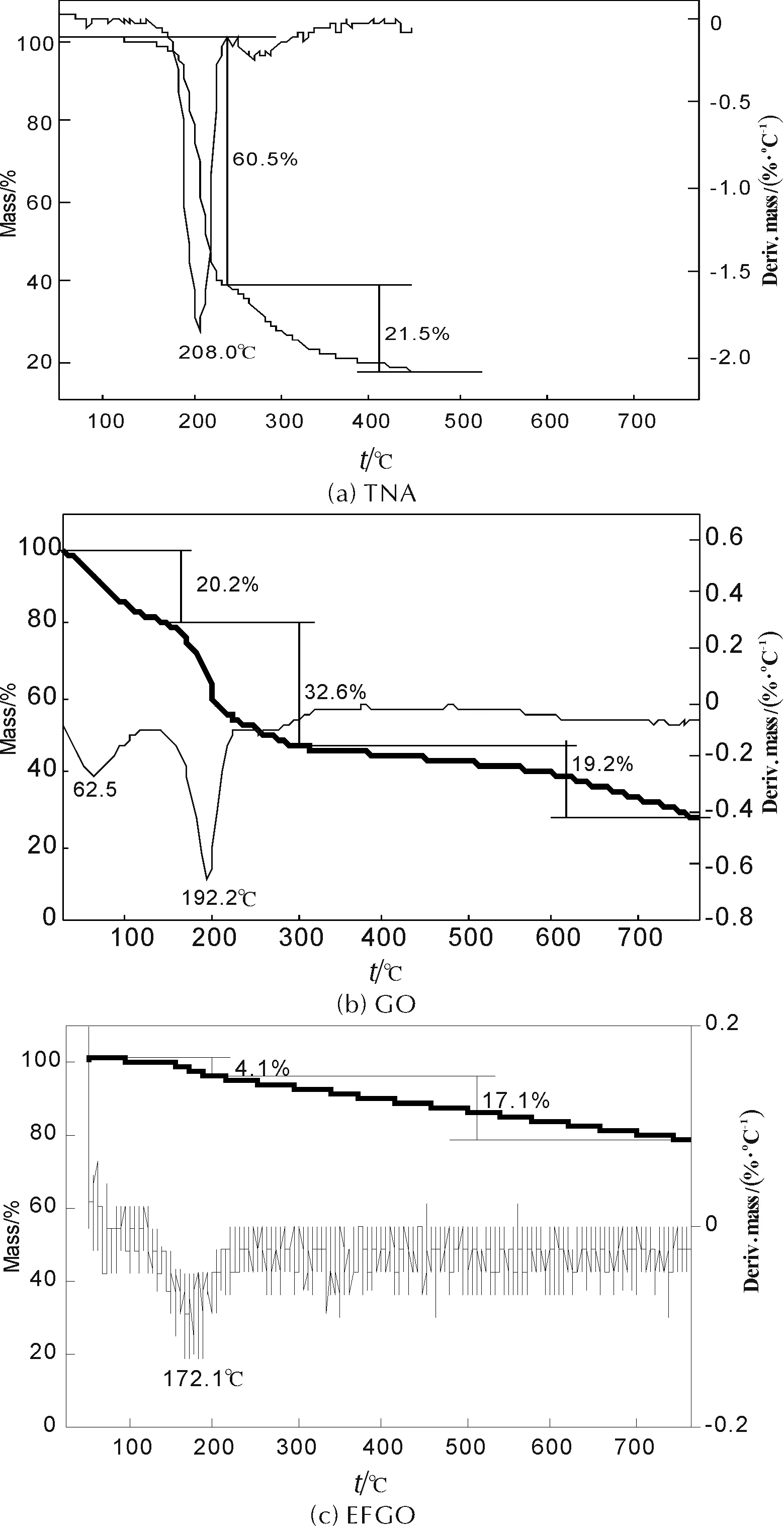
圖5 升溫速率10℃/min下TNA、GO和EFGO的TG-DTG曲線
由圖 5(a)可知,TNA熱穩定性較高,其熱失重過程可分為兩個階段:第一失重階段為劇烈失重過程,發生于50.0~236.5℃,失重60.5%,最大失重率發生在208.0℃;第二失重階段為緩慢失重過程,發生于236.5~450.0℃,失重21.5%。最終,TNA熱分解殘渣質量分數為18.0%。
由圖 5(b)可知,GO熱失重過程可分為3個階段[17]:第一失重階段為劇烈失重過程,發生于25.0~150.3℃,失重20.2%,最大失重率發生在62.5℃。該階段為GO脫水過程;第二失重階段為劇烈失重過程,發生于150.3~302.6℃,失重32.6%,最大失重率發生在192.2℃。該階段為GO表面、邊緣含氧官能團發生脫除、分解反應并生成H2O、CO和CO2過程;302.6~770.0℃為GO熱失重第三個階段,發生緩慢失重過程。在這個過程中,GO表面、邊緣殘留的含氧官能團進一步發生脫除、分解反應,最終生成rGO。最終,GO熱分解殘渣質量分數為28.0%。
由圖 5(c)可知,EFGO在143.8~770.0℃范圍內發生了非常緩慢的失重過程,可分為兩個階段:第一失重階段為劇烈失重過程,發生于143.8~204.0℃,失重4.1%,最大失重率發生在172.1℃;第二失重階段為緩慢失重過程,發生于204.0~770.0℃,失重17.1%。EFGO整個過程失重21.2%,相對于GO(72.0%)大大減少。最大失重率發生在約172.1℃,與GO第二階段最大失重率所對應的溫度接近,推測EFGO在172.1℃左右,其片層表面、邊緣的含氧官能團,以及含能官能團TNA發生脫除,分解和裂解反應。
以上結果表明,EFGO熱穩定相對于GO得到顯著提高,可能的原因為:TNA熱穩定性較高,將其通過共價鍵修飾至GO片層表面,TNA與GO平面產生π-π共軛效應,這種效應有利于提高EFGO熱穩定性。
2.3 GO和EFGO對AP熱分解的催化作用
為評價EFGO應用于AP基固體推進劑的可行性,分別對純AP、GO/AP和EFGO/AP進行DSC測試和分析,評價GO和EFGO對AP熱分解反應的催化性能。純AP、GO/AP和EFGO/AP的DSC曲線如圖 6所示。
由圖 6(a)可見,純AP熱分解分兩步進行[18]。其中,242.5℃左右為純AP相變溫度。264.3~345.1℃為純AP低溫分解階段(LTD),峰溫為330.2℃。345.1~441.8℃為純AP高溫分解階段(HTD),峰溫為432.5℃。純AP的LTD和HTD總的表觀分解放熱量為655 J/g。
由圖 6(b)可知,加入GO對AP相變過程沒有顯著的影響。加入GO后,AP在190℃左右出現放熱峰。此外,GO/AP熱分解行為相對純AP發生明顯的變化,但LTD和HTD的起始分解溫度和峰溫并沒有發生明顯的前移:GO質量分數分別為2.5%、5%和10%時,GO/AP LTD與HTD之間出現一個新的放熱峰,峰溫分別為358.9、365.3和355.5℃。LTD峰溫分別提前至318.5、313.9和308.5℃,相對純AP分別提前11.7、16.3和21.7℃。HTD峰溫相對純AP都沒有發生明顯的前移。GO質量分數分別為2.5%、5%和10%時,GO/AP LTD和HTD總的表觀分解放熱量分別增至995、1449和2024 J/g,相對純AP分別提高340、794和1369 J/g。上述結果表明,GO對AP熱分解反應的催化活性不高,與文獻結果一致[7-10]。
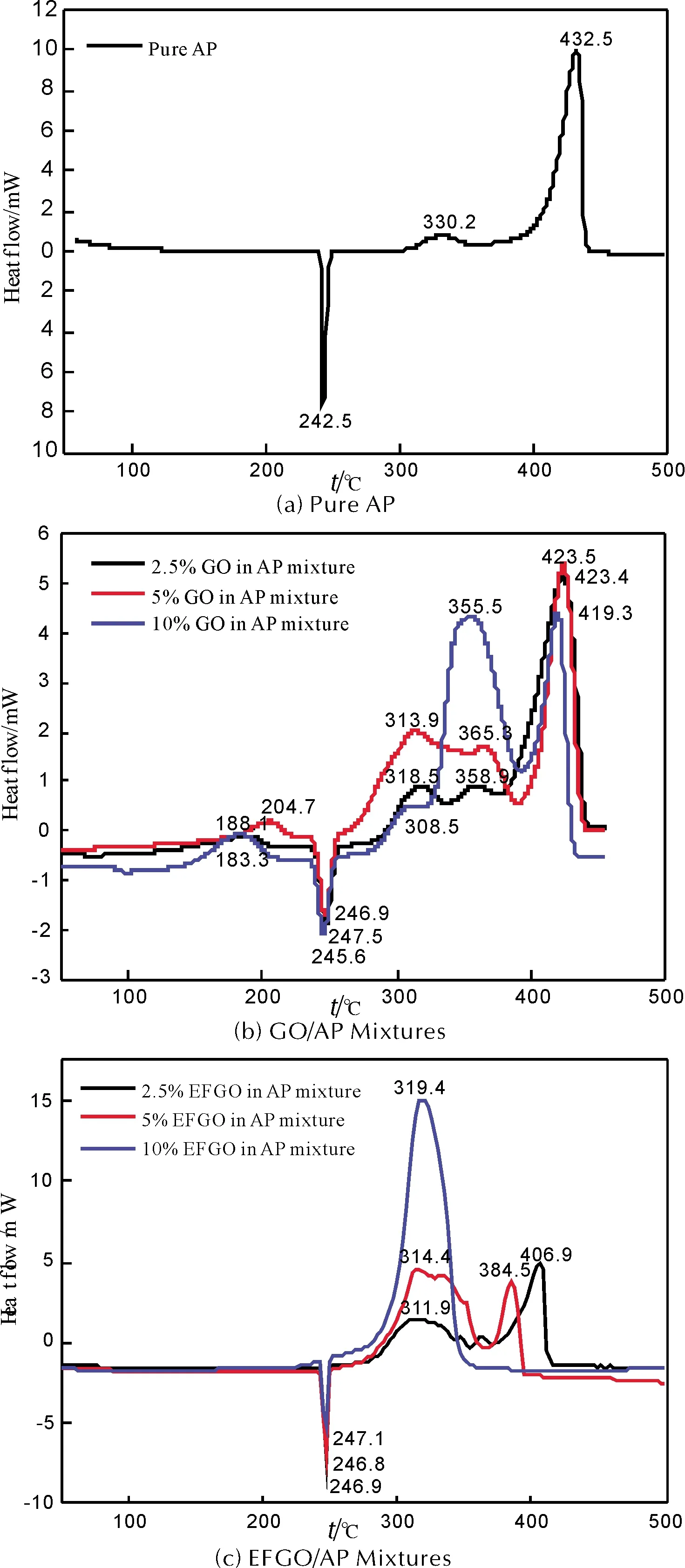
圖6 升溫速率10 ℃/min下純AP、GO/AP和EFGO/AP的DSC曲線
由圖 6(c)可知,在EFGO催化作用下,EFGO對AP相變溫度沒有明顯的影響。EFGO/AP熱分解行為相對純AP和GO/AP發生顯著的變化:LTD和HTD的起始分解溫度和峰溫發生明顯前移,表觀分解放熱量顯著增大。EFGO質量分數分別為2.5%和5%時,EFGO/AP熱分解反應仍然分LTD和HTD兩步進行。LTD和HTD峰溫分別提前至311.9、 314.4℃和406.9、384.5℃,相對純AP分別提前18.3、15.8℃和25.6、48.0℃。EFGO質量分數分別為2.5%和5%時,EFGO/AP LTD和HTD總的表觀分解放熱量分別增加至1026和2154J/g,相對純AP分別提高371和1499J/g。EFGO質量分數進一步提高至10%時,EFGO/AP熱分解行為發生根本性變化:EFGO/AP LTD與HTD完全合并,過渡階段完全消失。高溫熱分解階段的峰溫進一步提前至319.4℃,相對純AP提前113.1℃。表觀分解放熱量進一步增至3348J/g,相對純AP提高2693J/g。以上結果表明,將TNA通過共價鍵修飾至GO表面后,產物EFGO可使AP熱分解反應溫度明顯前移,表觀分解放熱量顯著增大,其催化活性相對GO得到顯著提高。
由純AP、GO/AP和EFGO/AP的DSC曲線和分析結果可知,GO對AP熱分解反應表現較低的催化活性,將TNA通過共價鍵修飾至GO表面后,產物EFGO催化活性相對GO得到顯著提高。對于純AP,LTD與HTD之間存在一個明顯的過渡階段,這是由于LTD的分解產物NH3大量吸附于AP表面,抑制AP進一步發生熱分解反應,導致LTD終止。當反應溫度進一步升高,吸附于AP表面的NH3開始在AP表面發生解吸,AP開始高溫分解反應,即進入HTD[18]。
對于GO/AP,在190℃左右出現放熱峰。由GO的TG-DTG曲線分析可推斷,在190℃左右,GO/AP混合物中GO表面、邊緣含氧官能團發生脫除、分解反應。隨著溫度的升高,GO表面、邊緣殘留的含氧官能團進一步發生脫除、分解反應,并生成rGO[17]。相對于GO,rGO的導電性和導熱性得到顯著提高,可促進AP LTD和HTD的電子轉移過程[19-20]。此外,rGO可與AP LTD和HTD的氧化性氣體產物發生氧化還原反應[21]。因此,GO對AP熱分解可表現一定的催化活性,使AP熱分解反應溫度略微提前,表觀分解放熱量明顯增大。
對于EFGO/AP,隨著EFGO含量的增加,LTD與HTD之間過渡階段的溫度區間逐漸變窄,直至完全消失。劉子如[12]研究“HMX與AP之間的相互作用”發現,HMX可加速AP熱分解,主要原因是HMX熱分解產物NO2可氧化AP的離解產物NH3,進而促進AP的高溫分解反應。為確定EFGO熱分解反應過程中氣相產物組成,對EFGO進行了TG-IR測試,結果如圖 7所示。
由圖 7可知,EFGO熱分解反應過程中氣相產物[18-21]主要是CO2(610~730和2280~2400 cm-1)、H2O(1550~1660和3650~3760 cm-1)和NO2(1610~1640 cm-1),以及少量NO(1890~1950 cm-1)和N2O(1230~1355 cm-1),證明EFGO熱分解反應過程中產生NO2。基于劉子如[12]的研究結果,GO/AP、EFGO/AP和EFGO熱分解反應的分析,以及EFGO的結構分析,可推斷:EFGO對AP熱分解反應的催化機制可能是EFGO分子中TNA與GO協同作用的結果。EFGO/AP熱分解反應過程中,一方面,EFGO片層表面的含能官能團TNA裂解并產生NO2,NO2可促進AP起始分解階段離解產物NH3的氧化反應;另一方面,EFGO片層表面、邊緣的含氧官能團在熱分解反應過程中發生脫除、分解反應,并生成rGO。NO2和rGO產生協同作用,可共同促進AP熱分解反應。因此,相對于GO,EFGO對AP熱分解表現較高的催化活性,使得AP熱分解反應溫度明顯提前,表觀分解放熱量顯著增大。隨著EFGO含量的增加,這種協同效應逐漸增強,EFGO/AP LTD與HTD之間過渡階段的溫度區間逐漸變窄,直至完全消失。
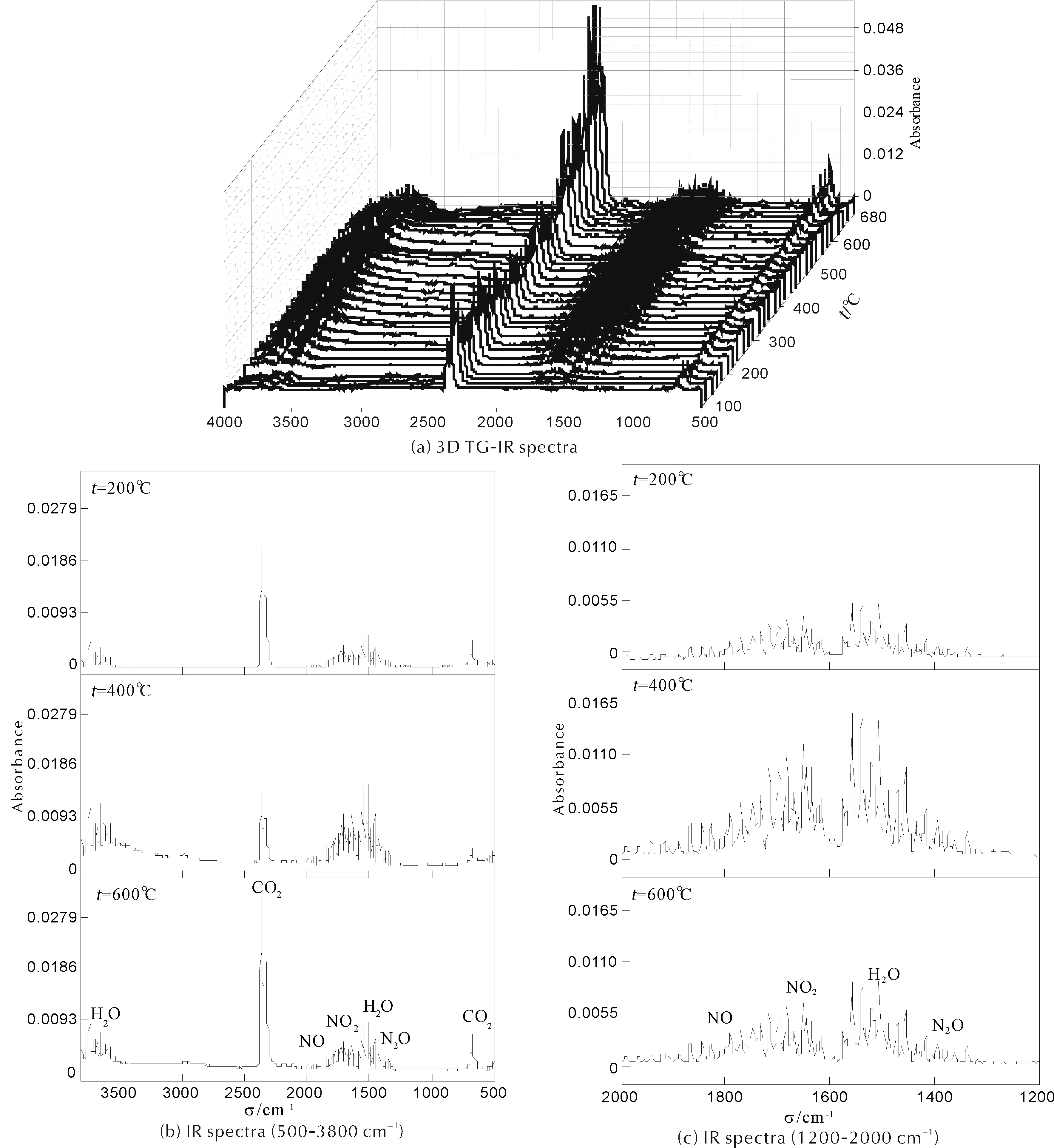
圖7 升溫速率10℃/min下EFGO熱分解反應氣相產物的紅外光譜圖
3 結 論
(1)TNA通過共價鍵修飾至GO片層表面,其中,TNA分子中—NH2與GO片層表面C—O—C發生共價鍵合,生成C—NH鍵。
(2)EFGO熱穩定性高,在143.8~770.0℃范圍內發生了非常緩慢的失重過程,最大失重率發生在約172.1℃,失重21.2%。
(3)EFGO催化活性相對GO得到顯著提高。EFGO質量分數10%時,可使AP的LTD和HTD完全合并,峰溫提前113.1℃,表觀分解放熱量增加2693J/g,對AP熱分解表現較高的催化活性。EFGO對AP熱分解反應的催化機制可能是EFGO分子中TNA與GO協同作用的結果。
(4)EFGO對AP熱分解反應的催化活性較高,將其作為非金屬含能催化劑應用于AP基固體推進劑,具有潛在的應用前景。對GO進行含能功能化修飾,可能是獲取新型高能非金屬燃燒催化劑的新途徑。
