山羊基因組與遺傳變異圖譜研究進展
2020-05-13 14:57:10李曉凱范一星喬賢張磊王鳳紅王志英王瑞軍張燕軍劉志紅王志新何利兵李金泉蘇蕊張家新
生物技術通報 2020年4期
關鍵詞:利用
李曉凱 范一星 喬賢 張磊 王鳳紅 王志英 王瑞軍,2,3 張燕軍,2,3 劉志紅,2,3 王志新 何利兵 李金泉,2,3 蘇蕊,2,3 張家新
(1. 內蒙古農業大學動物科學學院,呼和浩特 010018;2. 農業部肉羊遺傳育種重點實驗室,呼和浩特 010018;3. 內蒙古自治區山羊遺傳育種工程技術研究中心,呼和浩特 010018;4. 內蒙古金萊牧業科技有限責任公司,呼和浩特 010018)
基于考古學和遺傳學等方法的研究表明,家山羊是約在10 000 年前的新石器時代由西亞肥沃新月地帶的野山羊(Bezoars,Capra aegagrus)馴化而來,是最早馴化的反芻動物之一[1-2]。隨著人類的遷徙與演化,山羊是目前全球范圍內分布最廣泛的牲畜物種之一,主要用于生產肉、奶、皮和毛(絨)等農業生產資源[3-4]。據統計資料顯示,全世界范圍內共有10 億多只不同生產用途的山羊飼養在各種生態區內,超過90%的山羊分布在亞洲和非洲;其次是美洲、歐洲和大洋洲,包括肉用、乳用、皮毛用、絨毛用和普通山羊等不同生產用途的576 個山羊品種(http://www.fao.org/faostat/en/)[4-5]。山羊是發展中國家農牧民重要的家畜之一,但相對于奶牛、家豬、綿羊和家馬等經濟效益較高的牲畜品種,山羊的分子生物學研究和遺傳育種工作總體相對落后,嚴重阻礙了發展中國家貧困偏遠地區的經濟發展[6]。
隨著人類基因組計劃的實施與完成,單核苷多態性(Single-nucleotide polymorphism,SNP)因具有數量多,分布廣泛,易于快速、規模化篩查,便于基因分型等特點,已成為動物種質資源遺傳多樣性評估和基因功能定位研究的有力工具[7-9]。高通量測序技術的應用極大地促進了家畜基因組組裝和遺傳變異檢測的研究[10-11],如家牛[12]、家馬[13]、家豬[14]、山羊[15]和綿羊[16]等參考基因組組裝以及第一款家牛商業化芯片的研制[17]。2010 年,國際山羊基因組協會(International goat genome consortium,IGGC)成立,標志著高通量測序(Next generation sequencing,NGS)技術開始廣泛應用于山羊的基因組研究[18-19];2013 年完成了世界上首個山羊參考基因組草圖[15],并推出Goat SNP50K 磁珠芯片[20]和66K 目標捕獲芯片[21]。2017 年,Bickhart 等[22]組裝的近乎完整的參考基因組精細圖譜ARS1,為山羊功能基因的精細定位提供了更加可靠的基因組信息。通過對基因組的重測序、簡化基因組測序、外顯子測序和RNA-seq 等技術方法,與參考基因組(CHIR_1.0、CHIR_2.0 和ARS1)比對,獲得了大量的遺傳變異信息,為更全面的揭示山羊的遺傳多樣性、環境適應以及人工選擇反應提供了遺傳標記信息。因此,本文主要對山羊基因圖譜(遺傳圖譜、物理圖譜、轉錄圖譜與表達圖譜)以及分子遺傳變異信息的檢測進展進行了綜述,以期為進一步利用參考基因組信息和遺傳變異標記對山羊進行經濟性狀的遺傳基礎研究和分子育種提供參考。
1 山羊基因組圖譜
1.1 遺傳圖譜
1996 年,Vaiman 等[23]基于微衛星標記和共線性分析,利用12 個半同胞家系山羊(薩能奶山羊和阿爾卑斯山羊雜交種)構建得到了低分辨率的連鎖圖譜,并利用熒光原位雜交技術(Fluorescence in situ hybrid,FISH)確定了204 個微衛星標記;最終得到全長為2 300 cM 的連鎖圖譜,覆蓋了山羊基因組長度的80%。1998 年,Schibler 等[24-25]構建了山羊BAC 文庫,并通過ZOO-FISH 技術在山羊染色體上定位了202 個基因,同時在已有的山羊遺傳圖譜上增加了30 個微衛星標記,以此構建的細胞遺傳-遺傳連鎖合成圖含有307 個微衛星標記257 個基因,遺傳圖譜長度約2 737 cM,覆蓋了山羊基因組的88%。2005 年,Maddox[26]對綿羊和山羊的遺傳圖譜進行比較,結果顯示有218 個公共共有基因座,同時發現它們的同源基因座在圖中的位置很一致。
1.2 物理圖譜
1975 年,Goss 和Harris[27]共同創立了體細胞雜交技術,即輻射雜交(Radiation hybrid,RH)基因組作圖技術,其原理是用輻射來誘導染色體斷裂,并將輻射過的細胞與正常細胞進行雜交,獲得含有染色體片段的雜種細胞。隨后利用輻射雜交技術成功在人類、家屬、家牛等不同物種中構建了基因組長范圍的高分辨連續物理圖譜,極大了促進了人類、小鼠及不同家畜物種的基因組研究進展。Du 等[28-29]利用輻射雜種嵌板技術,首次構建了山羊全基因組輻射雜種圖譜(CHIRH5000),為標記密度最高的的輻射雜種圖譜。隨后,更多的標記定位在山羊細胞遺傳-遺傳連鎖合成圖,這些研究和相應建成的山羊圖譜數據(http://locus.jouy.inra.fr)加深對哺乳動物染色體進化的了解,加速反芻動物圖位克隆的研究[30]。
1.3 EST與轉錄表達圖譜
基因組中僅包括2%左右的序列為編碼蛋白質,表達序列標簽(Expressed sequence tags,ESTs)和RNA-seq 測序可以最有效率的進行基因識別。構建生物特定組織、器官或細胞的cDNA 文庫并進行大規模EST 測序和RNA-seq 測序分析,能直接獲得大量的功能基因結構及表達特征,并以此來構建各種組織器官的基因表達譜和對基因組結構和功能進行注釋。1996 年,Le Provost 等[31]首次采用泌乳期的山羊乳腺組織構建了cDNA 文庫,經過篩選對其中的435 個cDNA 克隆進行EST 測序,確認了77 個與山羊泌乳有關的基因或者蛋白。2000 年,Le Provost等[32]進一步采用圖位克隆的技術,結合EST 測序和細胞遺傳定位技術鑒定了25 個可能與產奶性狀有關的新的基因,其中6 個定位在牛的產奶QTL 區域。
RNA-seq 技術與生物信息學的快速發展,為理解基因組結構和基因功能奠定了基礎。Dong 等[15]對云南黑山羊不同組織(肝臟、心臟、肺、腎臟、脾臟、淋巴結、前腦皮層、肌肉、膀胱和卵巢)的mRNA 進行了轉錄組測序,為基因功能注釋奠定了堅實的基礎。不同組織、細胞的非編碼RNA 的檢測研究,如miRNA(乳腺[33]、皮膚毛囊[34-35]、卵巢[36]、垂體[37]、真皮乳頭細胞[38],以及背最長肌[39])、LncRNA(骨骼肌[40]、卵巢[41]和毛囊[42])等的分析研究,也為精細山羊基因組的功能結構、調控元件和基因功能注釋提供了數據支持。
1.4 參考基因組組裝
2010 年3 月,國際山羊基因組合作聯盟(International goat genome consortium,IGGC)在中國深圳正式成立,由中國科學院昆明動物所、深圳華大基因和內蒙古農業大學等10 多個國家的20 個科研機構或組織參與,旨在通過國際間的交流合作,加快山羊基因組圖譜構建、山羊遺傳多樣性、環境適應基礎和分子育種等方面的研究進展[43]。通過各個研究機構的合作努力和不同的技術方法,先后構建了家山羊參考基因組(CHIR_1.0、CHIR_2.0、ARS1 和CVASU_BBG_1.0)和野山羊參考基因組(CapAeg_1.0 和Caeg1),為加快山羊的分子生物學研究和今后的基因組選擇育種奠定了基礎。
1.4.1 家山羊參考基因組(CHIR_1.0 與CHIR_2.0)2013 年,Dong 等[15]利用Illumina 測序和光學圖譜(Optical mapping)技術以及Fosmid 和輻射雜種嵌板技術的數據對云南黑山羊進行基因組從頭組裝和染色體定位。對云南黑山羊母羊采用雙末端測序,構建了7 個不同大小片段文庫用于基因組測序,共產生191.5 Gb 高質量數據。首先,由17-kmer 推算和c-value 計算山羊的基因組大小,約為2.92 Gb。其次,利用SOAPdenovo 軟件經過初步組裝后的contig N50 為18 kb;scaffold N50 為2.2 Mb。最后,利用Fosmid 和Optical mapping 技術方法輔助構建Superscaffold,獲得最終的super-scaffold,獲得2.66 Gb 大小的參考基因組(CHIR_1.0),組裝出的基因組序列占預測基因組大小的92%(2.92 Gb),其Scaffolds N50 的大小為18 Mb,無法定位到染色體的superscaffold 歸類為chromosome U[15]。此外,利用RH技術對山羊第1 號染色體構建了高密度SNP 標記的輻射雜種圖譜,并與Optical mapping 數據組裝的長超級支架(Super-scaffold)進行了比對,成功證明了山羊序列的組裝質量的可靠性[29]。山羊基因組中含有大量重復序列,約占基因組42.2%。使用從頭注釋、基于人和牛的基因同源注釋和基因預測,總共注釋出山羊蛋白編碼基因有22 175 個,平均轉錄本長度為29 969 bp,CDS 平均長度為1 385 bp,每個基因平均含有8 個外顯子,每個外顯子的平均長度為168 bp,內含子平均長度為3 956 bp。隨后,研究人員進一步通過增加Illunima 測序數據,對參考基因組CHIR_1.0 進行了的一些修正,并利用輻射雜交技術修正了一些scaffold 的方向和順序以及掛載了CHIR_1.0未能成功掛載的scaffold[29]。通過一系列的組裝優化工作,最終獲得了山羊的基因組序列大小2.85 Gb,contig N50 的長度為29.87 kp,scaffold 的N50 長度為8.92 Mb,其中染色體的 GC 含量為 40.73%,;在使用CHIR_1.0 為模板掛載染色體后,同樣使用了野山羊染色體和綿羊染色體的作為模板掛載了剩余部分中未成功定位的scaffold,最終在CHIR_2.0 中的scaffold 序列中能成功掛載到山羊染色體上的序列占總序列的93.2%[44]。總的來說,相較于CHIR_1.0 版本的基因組,CHIR_2.0 在基因組完整性、功能注釋等方面都有較大的提升,極大地促進了山羊遺傳變異檢測和功能基因定位的研究 工作。
1.4.2 家山羊參考基因 組(ARS1) 2017 年,Bickhart 等[22]首先利用Illumina 的Goat SNP50K 芯片從96 頭山羊(6 個品種)中,篩選出基因型純和度最高的候選個體用來進行基因組從頭組裝(San clemente)。第一 步, 用Celera Assembler PacBio corrected Reads 流程對Pacbio 技術的465 個SMRT cell 產生的long-read,覆蓋深度達69X 的194 Gb基因組數據進行初步組裝,共獲得3 074 個contig(2.63 G),其中N50 為4.159 Mb。第二步,基于Irys optical mapping 技術對其雄性后代測序產生的256 Gb光學圖譜數據,并利用IrysView 軟件構建scaffold,組裝產生了842 個scaffold,其中,scaffold N50 為13.408 Mb(最長 的scaffold 為66.728 Mb),contig N50 為10.858 Mb。第三步,基于PacBio 和光學圖譜組裝的結果,構建Hi-C 文庫并物理方法打斷成300-500 bp 大小,雙末端(PE101),共產生115 Mb reads的數據量,調用Lachesis 軟件包,整合PacBio-Irys-PGA(PBIP),獲得Scaffold N50 為87.347 Mb 較為完美的組裝結果。第四步,利用Illumina 技術,構建PE251 測序,獲得23X 的基因組數據,用來進行一致校正和最后的補洞。最后,利用Kraken v0.10.5 去除有病毒和細菌污染的序列,去掉有NCBI vector污染的序列,獲得最終的2.924 Gb 大小的參考基因組圖譜ARS1,包含31 個scaffold,663 個gap 區和680 條contig。此外,利用6 個組織(大多和腦組織相關)RNA-seq 測序數據、13 個SRA 下載數據,用PASA 軟件將stringtie、cufflinks 和Trinity 分析結果整合在一起;用exonerate 和tblastn 軟件比對到幾個近緣物種的Ensembl 基因集上,獲得同源預測基因集;用Braker1 做Ab initio 預測;CHIR_1.0 版本的注釋基因集;最后,用EVM+PASA 把以上4 種數據整合成一個最終的基因集(設置的權重為RNAseq> cDNA/protein>ab initio gene predictions)。此版本基因組是目前組裝結果最好的山羊參考基因組,相應的組裝策略和技術為其他物種的參考基因組提供了參考,如最新獲得水牛基因組組裝就采用相似的 方法[45]。
1.4.3 家山羊參考基因組(CVASU_BBG_1.0) 2019年,Siddiki 等采用Illumina 測序平臺對孟加拉黑山羊進行深度為14X的150 bp 雙末端測序,利用ABySS v.2.1.5 組裝軟件初步獲得3 294 295 個contigs(最小contig 大小為200 bp)[46-47];進一步利用ABACAS v.1.3.1 組裝流程與參考基因組ARS1比較[48],進行從頭組裝基因組的排列、排序和定向,最終獲得了基因組大小為3.04 Gb 的孟加拉黑山羊參考基因組(CVASU_BBG_1.0);BUSCO 評估基因組的完整性為82.5%[49],基因注釋共發現了26 458個基因[50]。孟加拉黑山羊的基因組組裝結果為今后深入研究其種群遺傳結構、遺傳多樣性,評估該山羊品種的未來育種潛力奠定了堅實的基礎[51]。該研究中利用Illumina 短讀長數據進行初步組裝[47];隨后與參考基因組精細圖譜(ARS1)比較,利用ABACAS 等組裝進行基因組序列的排序和定向研究,為今后不同山羊品種的參考基因組組裝和進行山羊的泛基因組研究提供了可行性參考。
1.4.4 野山羊參考基因組(CapAeg_1.0) 2015 年,Dong 等[44]采用家 山羊CHIR_1.0 的DNA文庫構建方法對一只雄性野山羊進行測序,基于Illumina Hiseq 2000 測序平臺共獲得了381.50 Gb 大小的基因組數據;使用SOAPdenovo 軟件初步組裝獲得了野山羊基因組序列;隨后,基于野山羊與家山羊基因組的共線性關系,使用LASTZ 軟件與家山羊參考基因組比對信息,構建了野山羊常染色體基因組。為進一步構建野山羊Y 染色體基因組數據,首先利用BLAT 軟件將常染色體組裝中未錨定位置的Scaffolds與家牛Y 染色體(家牛Btau_4.6.1 的NC_016145.1染色體)參考基因組進行比對;反過來利用LASTZ軟件將家牛Y 染色體的contigs 比對到野山羊Scaffolds 上,通過過濾檢驗分析,最終獲得野山羊參考基因組CapAeg_1.0,其中contig N50 為18.97 Kb,scaffold N50 為2.06 Mb;Y 染色體大小為17.3 Mb,包含79 個錨定的scaffolds。為注釋野山羊基因組的蛋白編碼基因,采用了從頭預測,同源蛋白比對,轉錄組測序數據和序列表達標簽信息,注釋出了23 217 個基因;其中注釋到了57 個Y 染色體基因,包括11 個已知的雄性特有基因(Male specific region genes,MSY)。獲得了大量的遺傳變異信息,其中揭示了ASIP基因的拷貝數變異與家山羊的被毛變化相關。
到目前為止,獲得的從頭組裝的參考基因組共有4 個品種的個體,其中以ARS1 組裝注釋結果最好(不同參考基因組詳細信息見表1),這些組裝到的基因組在一定上促進了山羊泛基因組的研究,為揭示基因組水平大規模的變異奠定的基因組水平的數據基礎。
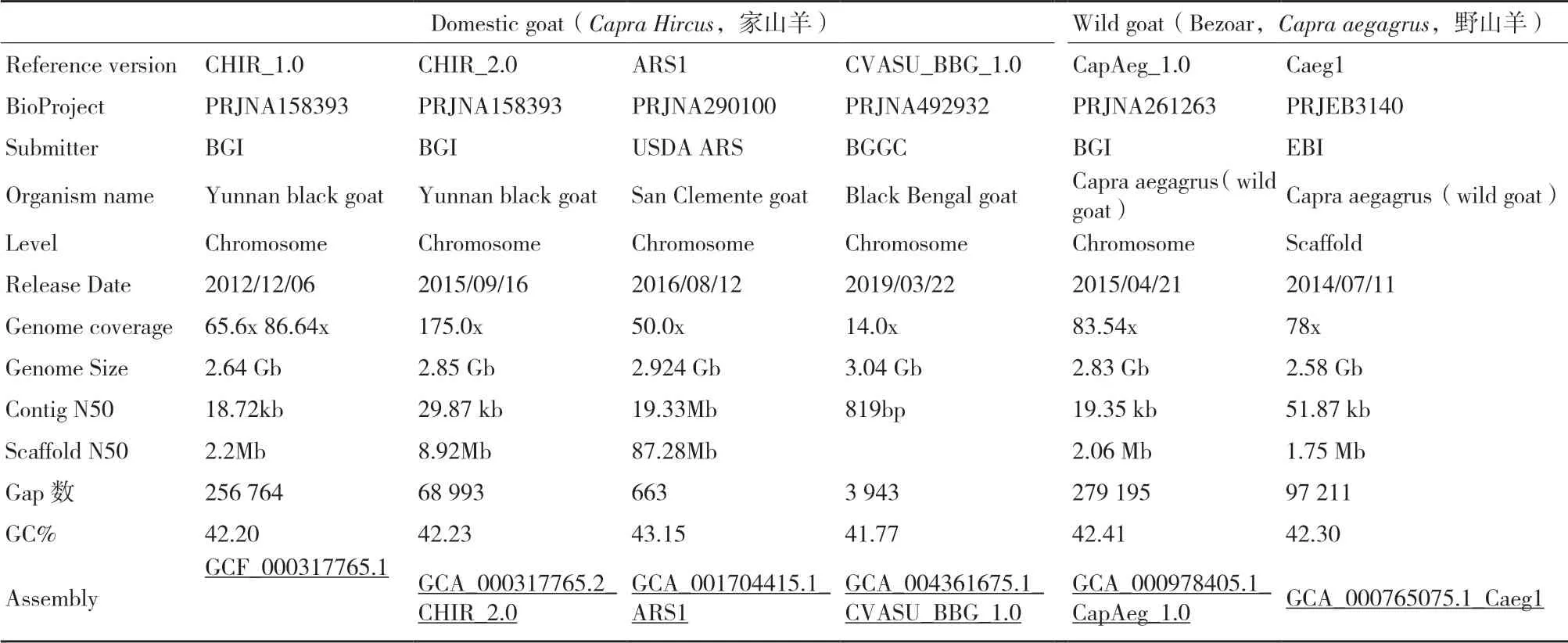
表1 不同山羊基因組de novo 組裝版本信息
2 全基因組變異圖譜
隨著測序技術的不斷成熟及測序成本的不斷降低,利用高通量測序技術檢測山羊全基因組水平的遺傳變異逐漸成為可能。此外,隨著研究對象樣本量和品種數的增加,山羊遺傳變異的信息也逐漸增加和豐富,極大了加深了我們對的山羊遺傳多樣性和環境適應性的理解(http://www.genome.gov/sequencingcosts/)。根據遺傳變異形成機制、存在形式以及對基因組結構和表型的影響,可分為以下類型,即單核苷酸多態性、1-50 bp 的小片段的插入或缺失、50 bp 以上的拷貝數變異以及由位置變化引起的易位或倒位等,詳細信息如圖1 所示[52]。
2010 年,Fontanesi 等[53]利用牛- 山羊間的微陣列比較基因組雜交(Array comparative genome hybridization,aCGH)技術,首次對山羊基因組拷貝數進行了檢測研究,共發現了161 個CNVs 變異。Liu 等[54]利用CaprineSNP50 芯片和PennCNV 軟件對ADAPTmap 項目產生的基因組數據進行CNV 分布分析,從50 個山羊品種的1 023 個個體中共獲得包含6 286個CNVs的978個區域,約262 Mb(8.96%)。基于SNP 芯片檢測CNV 的研究,擴展了SNP 芯片的應用范圍,加深了對CNV 變異在家畜遺傳多樣性和經濟性狀差異的理解,但因為SNP 芯片的敏感性等原因,其準確性和可靠性需要進一步驗證。
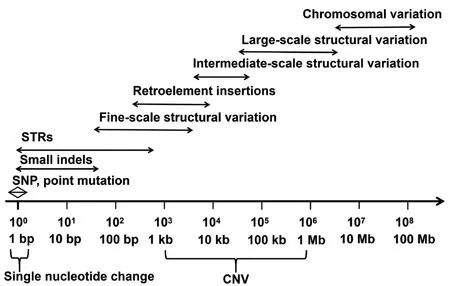
圖1 人類基因組的變異圖譜[52]
基于全基因組個體重測序的方法,Tosser-Klopp[20]、Dong[44]、Benjelloun[55]、Zhang[56]、Florian[55,57]、Li[58]、Lee[59]、Kim[60]和Cao[61]等對阿爾卑斯山羊、克里奧山羊、Katjang 山羊、Savanna 山羊、薩能奶山羊、波爾山羊、澳大利亞野化山羊、澳大利亞絨山羊、野山羊、摩洛哥山羊、遼寧絨山羊、內蒙古絨山羊、雷州山羊、韓國黑山羊、努比亞山羊和云嶺黑山羊等進行了2.7-30X不同深度的全基因組測序;采用全基因組混合池測序方法,Lai[62]、Zhang[63-65]、E[65-66]和Wang[67]等通過對嶗山奶山羊、大足黑山羊、太行黑山羊、西藏山羊、內蒙古絨山羊、陜北絨山羊、安哥拉山羊、薩能奶山羊、波爾山羊和貴州小山羊等進行了10-30X的混合池測序;基于簡化基因組測序方法,Song 等[68]對西藏班戈山羊和日土山羊、柴達木山羊、南疆絨山羊、內蒙古絨山羊二狼山型及遼寧絨山羊)不同個體進行了外顯子測序;Wang 等[69]利用RNAseq 技術對內蒙古絨山羊阿爾巴斯型進行了遺傳變異檢測分析。通過與參考基因組比對(CHIR_1.0、CHIR_2.0 和ARS1),檢測出大量的SNP、Indel 和CNV 等遺傳變異數據,為今后山羊分子遺傳標記的開發和利用以及遺傳資源保護奠定了堅實的基礎。
目前,隨著山羊分子生物學的不斷發展及對家畜分子育種的重視,許多研究機構對山羊的環境適應性和表型多樣性等方面進行了不同程度的研究,詳細信息見表2。因為測序項目實施的時間不同,所用到的山羊參考基因組信息有所不同,導致山羊遺傳變異在基因組上位置信息有所差異,為統一山羊基因組變異的相對位置,國際山羊基因組聯盟首先對Goat SNP50K 芯片的SNP 位置信息與ARS1 進行了比較和校正。由于NCBI 在2017 年逐漸停止對dbSNP 和dbVar 中的所有非人類生物的支持,目前山羊等物種的基因組變異數據存儲在Ensemble 數據庫 中(ftp://ftp.ensembl.org/pub/release-97/variation/gvf/capra_hircus/)。截止到2019 年5 月8 日,以參考基因組ARS1 版本的作為參考構建的遺傳變異信息,主要包括33 996 708 個SNP 和Indel,而CNV和SV 等的變異信息目前尚未公布。
3 遺傳變異信息的利用
山羊全基因組重測序研究的主要目標就是通過生物信息學方法檢測不同品種特有的選擇信號特征,揭示不同品種特異性的遺傳基礎;其次是構建不同品種的全基因組單倍型圖譜,為今后利用低密度芯片進行基因型填充、增加基因組信息的可利用率做基礎數據支持;再次是利用全基因組水平的遺傳變異信息,針對不同的研究群體和目標對SNPs 等遺傳變異信息進行過濾和篩選,進而開發不同密度的SNP 分型芯片。目前,利用不同品種的基因組遺傳變異信息,已經成功設計出了Goat SNP50K 芯片[20]和66K 目標捕獲芯片[21]。
基于全基因組重測序數據,在山羊的高海拔環境適應(EPAS1、EDNRA、SIRT1、PASK、PTPRZ1、NPC1L1和RYR1)[68]、脂肪代謝(ACSL1、LRP1、PLIN4、FASN)、絨用性狀(FGF5、PRDM6)[56,58,67]、被毛顏 色(KITLG、MC1R、ASIP、ATRN、GNAQ、
HELLS、MUTED、OSTM1、TRPM7、VPS33A、Ada-mts20,MITF、OCA2、SLC7A11和AHCY)[44,55,57]、 神經系統發育(ADRA2A、FXR2、HTR3A、CACNA1、CCHD5、ULK1、TMEM132A、SYNDIG1、ERC2和GABRB2)[44,56]、繁殖性狀(NR6A1、STK3、IGF2-BP2、NPTX1、ANKRD17、DPYD、CLRB、PPP3CA,PLCB1,STK3 and HMGA2,PRP1、PRP6、CCNB2、A R、ADCY1、DNMT3 B、SMAD2、AMHR2、ERBB2、FGFR1,MAP3K12、SETDB2、CDH26和THEM4)[62,64-65]、體尺性 狀(NR6A1、TNFSF13、STIM1、IGF1R)[44,56]、肉用性 狀(GDF5、LRP4、HMGXB3、SLC26A2、goat_GLEAN 10018710、SLC-35A3、HIAT1、SASS6和GOAT_ENSBTAP00000044-216)[56]、疾病抗性(HTT、CCR3)[55,59]、生長性狀(CCKAR、IGF1R、MYADM)[44]、免疫系統(ABCC4、PRAME、CD163L1、KIR3DL1、CFH和TRIM5)[44]、精子發生(PRAME)[44]和乳用性 狀(BTN1A1、RSRC1、SHOX2、VPS13A、VPS13B、VPS13C和RPL3)[44,56]等遺傳基礎的解析方面取得了眾多研究成果。
4 展望
目前,山羊重要經濟性狀遺傳基礎的研究正在由候選基因、單一性狀的方法向全基因組水平、多性狀和多組學等聯合分析的方法進行轉變。高通量測序技術的進一步發展和新的分析方法的不斷涌現,加快了研究人員研究、挖掘全基因組范圍內山羊的遺傳多樣性信息及經濟性狀相關的分子基礎,如Guan 等[71]基于共享基因組數據分析山羊酪蛋白基因家族變異的起源與演化過程。山羊大多數經濟性狀屬于數量性狀,遺傳因素如單堿基突變(SNP)、插入缺失(Indel)、結構變異(SV)和表觀遺傳修飾調控(甲基化修飾、組蛋白修飾和非編碼RNA 調控)以及環境和營養因素等均會影響到山羊的表型性狀和生產性能。為揭示復雜性狀的遺傳基礎和調控機制,高通量技術下的研究方法主要包括對不同組織器官的差異基因表達的RNA-seq 分析、基于不同品種雜交個體的等位基因特異性表達分析、基于全基因組重測序技術的選擇性清除分析、復雜性狀基因定位的全基因組關聯分析、表觀遺傳調控組蛋白修飾和甲基化分析以及非編碼RNA 調控的研究以及逐漸在上述技術方法基礎上衍生的多組學方法,如RNA-seq+GWAS、WGS+GWAS、eGWAS和BSA+RNA-seq 等聯合分析進行精確定位的研究方法[71-73]。通過合理的選擇研究對象、構建理想的試驗群體并適當的組學技術,借助公共數據庫基因組信息和生物信息學方法挖掘其潛在的與生產性狀相關的基因或基因組區域、影響效應和調控互作機制將是今后的研究重點,也對推動山羊分子育種和基因組選擇研究工作具有重要的理論和實踐意義。

表2 國際山羊遺傳資源與研究機構相關網站
猜你喜歡
中等數學(2022年2期)2022-06-05 07:10:50
中學生數理化·七年級數學人教版(2021年11期)2021-12-06 05:38:48
中學生數理化(高中版.高考數學)(2021年6期)2021-07-28 06:19:08
小學生學習指導(低年級)(2020年6期)2020-07-25 02:31:36
小學生學習指導(低年級)(2019年11期)2019-11-25 07:31:44
小學生學習指導(低年級)(2018年9期)2018-09-26 05:59:44
瘋狂英語·新讀寫(2018年2期)2018-09-07 09:32:10
數學小靈通·3-4年級(2017年6期)2017-06-22 11:28:50
工業設計(2016年5期)2016-05-04 04:00:33
河北遙感(2015年4期)2015-07-18 11:05:06
