綠木霉(Trichoderma virens)T23甲基轉移酶基因 gliN-T對膠毒素合成的調控研究
2020-06-04 01:37:48華麗霞蔣秋平曾華蘭葉鵬盛王明娟楊曉丫何曉敏
中國農業大學學報 2020年6期
關鍵詞:檢測
華麗霞 孫 佩 蔣秋平 曾華蘭* 葉鵬盛 何 煉 曾 靜 王明娟 張 敏 羅 飛 楊曉丫 何曉敏 劉 勇
(1.四川省農業科學院 經濟作物育種栽培研究所,四川 成都 610300;2.農業農村部西南作物有害生物綜合治理重點實驗室,四川 成都 610066)
木霉菌(Trichodermaspp.)是研究和利用最廣泛的植物病害生防真菌。已有研究表明木霉菌對黃瓜枯萎病病菌、辣椒疫霉病菌、棉花立枯絲核菌等多種植物病原真菌具有顯著拮抗效果,抑菌率在80%以上[1-2];木霉菌能增強作物根系活力,對作物的生長具有促進作用[3-5]。因此,木霉菌在作物病害生物防治、農藥減施增效方面具有重要的研究、開發及應用價值。膠霉毒素(Gliotoxin),簡稱膠毒素,是一種真菌次生代謝產物,因其對細菌、放線菌以及真菌等具有顯著的拮抗作用,已被開發成抗生素或化療藥物[6]。木霉屬中的部分菌種可產生膠毒素,該物質對病毒、細菌等均有抑制效果[7-8]。
膠毒素合成的分子機制在煙曲霉中研究較為深入:在煙曲霉基因組調控膠毒素合成的基因簇即gli基因簇中包含了12 個基因(gliA,gliC,gliF,gliG,gliK,gliM,gliN,gliJ,gliI,gliH,gliZ,gliT)共同調控著煙曲霉膠毒素的合成[9],目前,簇內gliP、gliC、gliZ、gliA、gliT在煙曲霉膠毒素合成途徑中的作用已經較為清晰[10~14]。已有研究表明綠木霉菌(Trichodermavirens)中的部分菌株可產生膠毒素,并通過脈沖標記明確了綠木霉菌液體培養過程中膠毒素含量的動態變化情況[15],但是綠木霉菌膠毒素合成分子調控機制方面的研究遠遠滯后于煙曲霉膠毒素的研究。雖然Vargas等[16]對T.virens菌株Gv29-8中的gliP基因進行基因敲除,明確了該基因與綠木霉膠毒素的合成有關,然而,在分子水平上深入剖析綠木霉菌膠毒素合成的分子調控機制仍需要進行大量的科研工作。因此,開展綠木霉菌膠毒素合成基因的克隆及功能研究,有利于對膠毒素合成代謝的分子調控機制進行深入研究,有利于從分子水平上闡明木霉生防菌的拮抗機理,為進一步開發和應用提供重要的理論基礎。
本研究室前期通過生物信息學方法,發現綠木霉菌T23基因組中存在1 個與煙曲霉膠毒素合成基因簇有同源性的基因簇,簇內有8 個候選基因,分別命名為gliP-T、gliC-T、gliN-T、gliK-T、gliI-T、gliG-T、gliF-T、gliM-T[17]。為進一步研究綠木霉菌膠毒素合成的分子調控機制,本研究擬以綠木霉菌T23為研究對象,通過改良的農桿菌介導的遺傳轉化(Agrobacteriumtumefaciens-mediated transformation, ATMT)技術,對T23膠毒素合成基因簇內編碼帶甲基轉移酶結構蛋白的gliN-T基因進行敲除,分析基因敲除突變體中膠毒素含量變化情況,明確gliN-T對膠毒素合成的調控作用,為后續的膠毒素代謝調控機制的深入剖析提供重要的理論基礎,同時為膠毒素的進一步開發利用提供參考信息。
1 材料與方法
1.1 供試材料
試驗菌為生防綠木霉菌T23,由本研究室從川產道地中藥材根際土樣中分離得到[18]。
細胞壁裂解酶Glucanex(Sigma-Aldrich,L1412,美國);E.Z.N.A.?Fungal DNA Kit(Omega Biotek,D3390=01,美國);膠毒素標準品(1 mg,Lot number: 1D0E12, 純度>99%,青島普瑞邦生物工程有限公司);高效硅膠GF254預制板(青島海洋化工有限公司);試驗所需引物的合成及測序均由生工生物工程(上海)股份有限公司完成。
1.2 儀器設備及試劑
高效液相色譜1200型,配紫外檢測器G1314B(安捷倫科技有限公司,美國);C18反相色譜柱(4.6 mm×250 mm, 5 μm, Pribolab, Singapore);色譜純甲醇(美國Fisher);
1.3 培養基及相關試劑
馬鈴薯葡萄糖瓊脂培養基(Potato dextrose agar, PDA)用于綠木霉菌T23的培養;LB培養基用于大腸桿菌及農桿菌的培養;IM液體誘導培養基用于ATMT誘導轉化[19],121 ℃高溫高壓滅菌后,加入終濃度為200 μmol/L的乙酰丁香酮;共培養基用于農桿菌和分生孢子的共培養,其配方在IM培養基基礎上,每升共培養基只需加入葡萄糖1 g,加入瓊脂粉 15 g,121 ℃高溫高壓滅菌后加入終濃度為 200 μmol/L 的乙酰丁香酮,倒平板待用。
篩選培養基即PDA培養基高溫高壓滅菌后,加入終濃度為150 μg/mL的潮霉素B后倒平板待用。
1.4 基因敲除載體的構建及轉化子的分子鑒定
1.4.1DNA 提取
將供試綠木霉菌株T23活化于PDA平板培養基上,培養2~3 d后,用接種環挑取新鮮菌絲接種到馬鈴薯葡萄糖液體培養基(Potato dextrose broth, PDB)中,28 ℃振蕩培養2~3 d,待菌絲體生長茂盛的時候,用滅菌紗布和濾紙過濾收集菌絲體,用紙巾盡量吸干菌絲體中的水分后置于滅菌研缽中,加入液氮研磨至粉末狀,然后用E.Z.N.A.?Fungal DNA Kit(Omega Biotek,D3390=01,美國)進行基因組DNA的提取,具體操作依照說明書進行。
1.4.2基因敲除載體構建及轉化子的分子鑒定
按照同源敲除的原理構建基因敲除載體(圖1):分別以gliN-T的5′端1 016 bp (LF)及3′端 892 bp(RF)作為同源臂;以構巢曲霉啟動子PtrpC啟動的潮霉素基因(hph),即潮霉素表達盒作為基因替換片段,按照LF::PtrpC-hph::RF的順序,組裝到攜帶有CaMV poly(A)終止子元件的基因敲除載體pGKO2中。根據PtrpC-hph序列設計正反方向引物,即hyp-F與hyp-R,用于轉化子潮霉素盒的檢測;同時根據LF序列設計正向引物gliN-F,根據RF序列設計反向引物gliN-R,用于同源重組轉化子的分子鑒定。引物信息見表1。
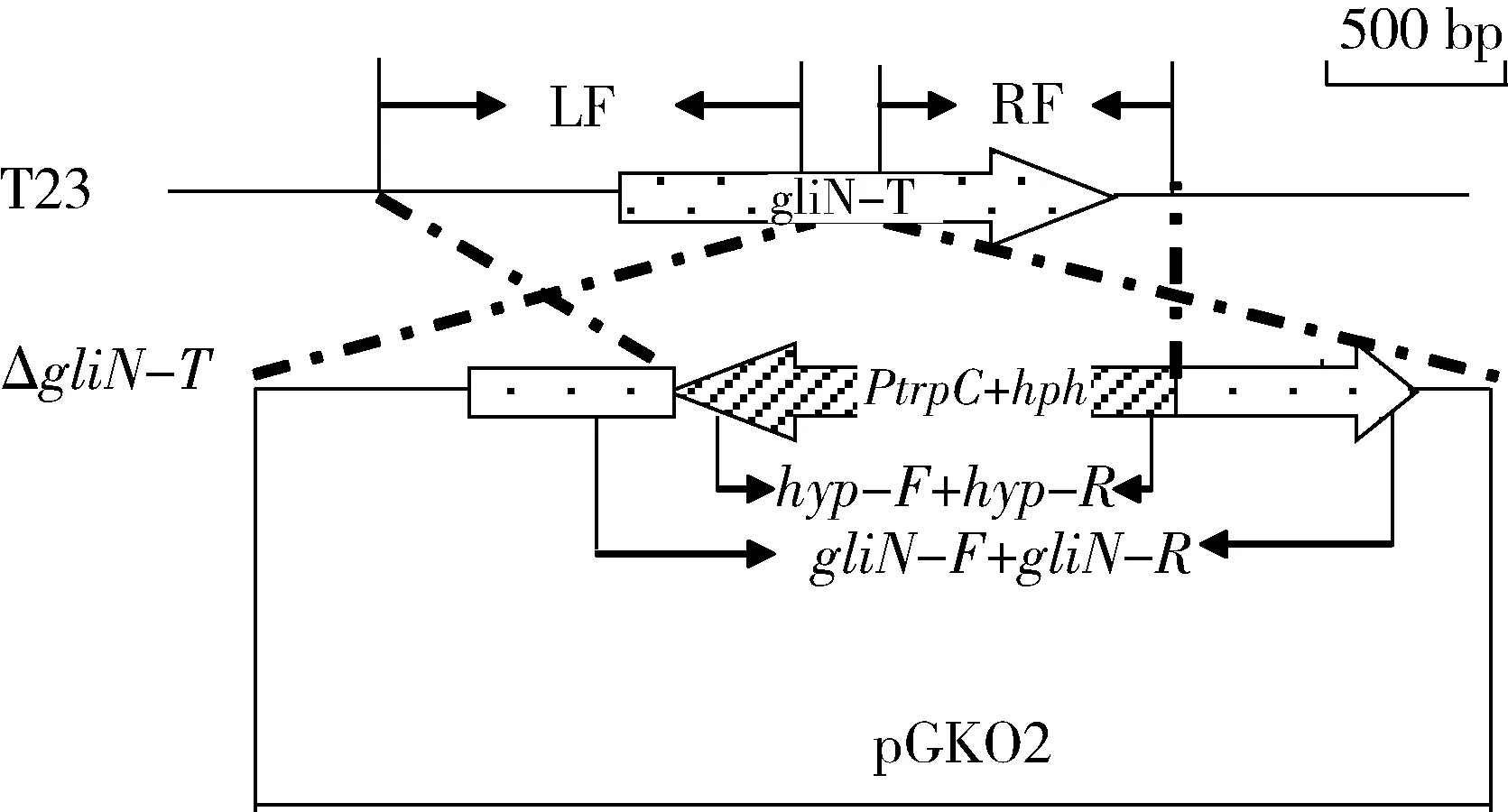
圖1 基因敲除同源置換原理
Fig.1 Gene knockout strategy based on homologous recombination

表1 本研究所用引物Table 1 Primers used in this study
注:引物序列5′末端的小寫字母為無縫拼接所需的同源重組序列;*,野生型和突變體擴增片段大小。
Note: Lowercase letters on the 5′-end present the homology of the adjacent fragment for vector construction using seamless cloning; *, length of PCR fragments of wild type and mutant.
1.5 農桿菌介導的遺傳轉化技術的改良
1)取攜帶有基因敲除構建體的陽性農桿菌C58C1在LB培養基上劃線,28 ℃培養36 h,挑取單菌落到帶抗生素的LB液體培養基中,28 ℃振蕩培養過夜,離心收集菌體,隨后用IM誘導培養基(含終濃度為200 μmol/L的乙酰丁香酮)重懸浮菌體并調節OD600至0.2~0.3,然后28 ℃振蕩培養6 h,備用。
2)綠木霉分生孢子懸浮液的制備:在PDA培養基上活化綠木霉菌株T23,28 ℃培養4 d左右至產孢,用滅菌水輕輕地刮洗菌面,3 層擦鏡紙過濾菌絲,4 000 r/m離心5~10 min收集分生孢子。倒掉上清,加入細胞壁裂解酶Glucanex(終濃度為15 mg/mL,溶于0.7 mol/L NaCl溶液),30 ℃ 80 r/m 振蕩培養2.5~3.0 h,4 000 r/m離心,用0.7 mol/L NaCl溶液清洗1~2 遍,3 層擦鏡紙過濾收集分生孢子液,調節分生孢子濃度至106個/mL。
3)取等體積(200 μL)的農桿菌菌液及孢子懸浮液進行混合后均勻涂布于共培養平板培養基中,22 ℃ 暗培養3 d。隨后,在共培養好的平板上倒1 層含潮霉素(終濃度150 μg/mL)及頭孢抗生素(終濃度400 μg/mL)的PDA篩選培養基,28 ℃培養 5 d 左右,挑取能在篩選培養基上生長的轉化子到新的篩選培養基中,進行潮霉素穩定性鑒定。
1.6 基因表達分析
參照已有報道[17]提取野生型T23和基因敲除突變體ΔgliN-T的總RNA并進行反轉錄,合成cDNA第一鏈。在gliN-T基因外顯子區域設計引物,在轉錄組水平上檢測gliN-T在突變體中的表達情況。引物詳細信息見表1。
1.7 膠毒素的提取及檢測
1.7.1薄層色譜分析
將供試菌株接種到100 mL PDB培養基中,28 ℃ 震蕩培養48 h,用孔徑為0.22 μm的濾膜過濾培養液,取20 mL濾液,加入等體積的乙酸乙酯萃取2 次,合并萃取液,置于通風櫥中揮干得到待測樣品后用2 mL甲醇溶解作為薄層鑒別供試品。取膠毒素對照品1 mg,加入2 mL甲醇溶液,超聲5 min,得到濃度為0.5 mg/mL的膠毒素標準品溶液,作為薄層分析的對照品溶液。以高效硅膠GF254預制板作為薄層板,以正己烷-乙酸乙酯體積比為6∶6作為展開劑,在254 nm的紫外光下觀察薄層板的展開情況,隨后用碘蒸汽對薄層板進行顯色。
1.7.2高效液相色譜檢測
挑取新鮮菌絲接種至200 mLPDB培養基中, 28 ℃中震蕩培養48 h后取2 mL培養液并用0.22 μm 微孔濾膜過濾至進樣瓶中待用。按照以下的色譜條件對培養液中的膠毒素進行檢測:以水為流動相A,甲醇為流動相C,按照50%水加50%甲醇進行等度洗脫;流速:1 mL/min;柱溫:30 ℃;進樣量:20 μL;檢測波長:254 nm。
2 結果與分析
2.1 通過改良的ATMT技術獲得gliN-T基因敲除突變體
在細胞壁溶解酶Glucanex處理試驗前,共進行了3 批次的遺傳轉化試驗共篩選到3 個轉化子。分子鑒定結果表明這3 個轉化子均為異位整合轉化子。在分生孢子液中添加終濃度為15 mg/mL的細胞壁溶解酶Glucanex進行處理后,篩選得到22 個轉化子,潮霉素基因檢測陽性率為100%,基因特異引物鑒定結果見圖2:在22 個轉化子中:有3 個是同源重組轉化子,即成功敲除gliN-T的基因敲除突變體ΔgliN-T;有2 個轉化子檢測到潮霉素基因,但檢測不到同源敲除的條帶,為非正常轉化子;其余17 個是異位整合轉化子。對溶解酶處理前后的試驗結果進行比較分析,發現綠木霉菌T23分生孢子經細胞壁溶解酶Glucanex處理后,平均每毫升分生孢子液的轉化子數從0.25 個提升到10.00 個,轉化效率大大提高(表2)。
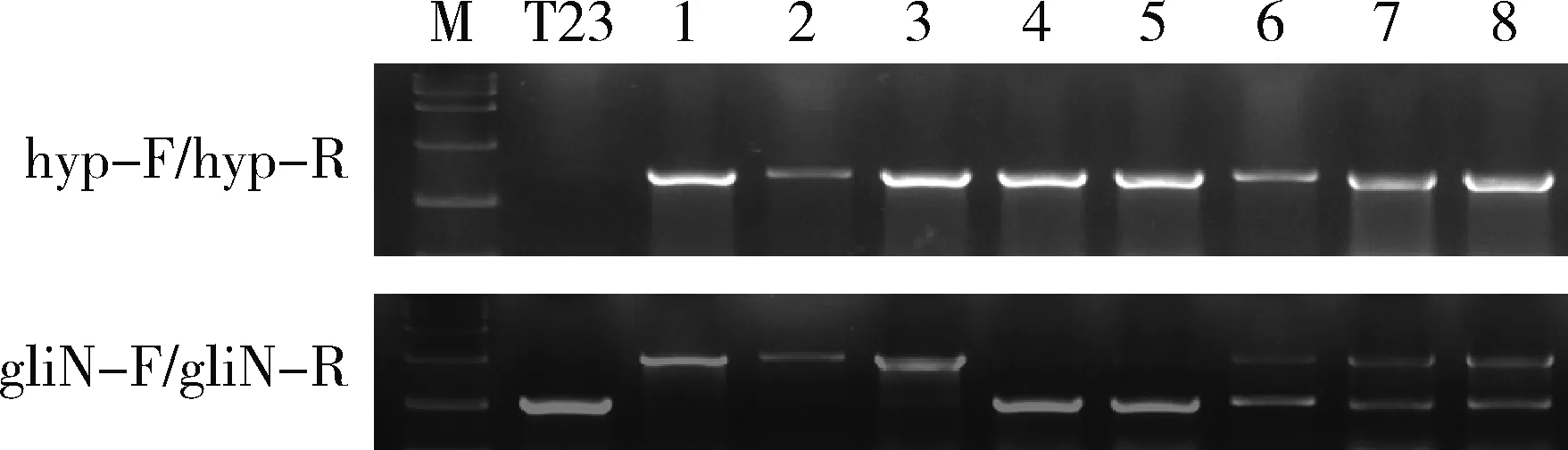
M:DL15000;T23:野生型T23 DNA擴增產物;1~8:基因敲除轉化子DNA擴增產物(1~3為同源重組陽性轉化子,4~5為非正常轉化子,6~8為異位整合轉化子)。 M: DL15000; T23: amplicon from wildtype T23 gDNA; 1-8 are amplicons from transformants gDNA: 1-3 represent homologous transformants; 4-5 are unusual transformants; 6-8 are ectopic transformants.
圖2 Glucanex處理后獲得的轉化子的分子鑒定
Fig.2 Molecular identification for transformants after glucanex treatment
2.2 gliN-T在基因敲除突變體中的表達分析
對野生型T23及基因敲除突變體ΔgliN-T中的gliN-T進行RT-PCR表達分析,結果表明:在野生型T23基因組及cDNA中均檢測到gliN-T的表達; 在轉錄水平上,在基因敲除突變體ΔgliN-T中檢測不到gliN-T的表達(圖3),意味著gliN-T基因被成功敲除。

表2 Glucanex處理前后木霉菌T23 ATMT轉化效果比較Table 2 Comparison of transformant efficiency before and after Glucanex treated for T23
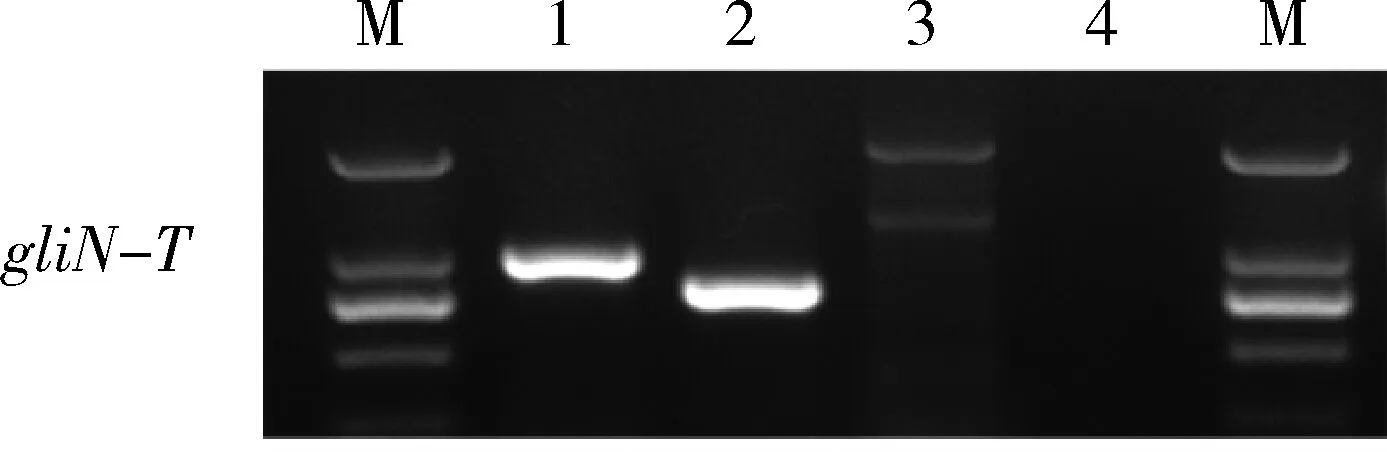
M:DL2000;1:T23 gDNA 擴增產物;2:T23 cDNA 擴增產品;3:ΔgliN-TcDNA 擴增產物;4:陰性對照,以H2O為模板的擴增產物。 M, DL2000; 1, amplicon from T23 gDNA; 2, amplicon from T23 cDNA; 3, amplicon fromΔgliN-TcDNA; 4, amplicon from H2O as negative control.
圖3gliN-T在基因敲除突變體ΔgliN-T中的表達分析
Fig.3 Expression analysis forgliN-Tin gene knockout mutantΔgliN-T
2.3 gliN-T基因敲除突變體膠毒素代謝產物的檢測
對野生型與基因敲除突變體ΔgliN-T的48 h的培養液進行薄層色譜分析,膠毒素標準品上樣量為5 μL,供試樣品上樣量為10 μL。薄層色譜分析結果表明,在野生型T23 48 h培養液中可檢測到膠毒素(圖4,泳道2和4);而相同培養條件下,基因敲除突變體ΔgliN-T的培養液則無法檢測到膠毒素(圖4,泳道3)。
為了進一步明確膠毒素的分泌是否與菌絲生長時期有關,本研究對不同培養時期(24、48、72和96 h)的野生型T23及基因敲除突變體ΔgliN-T的菌絲培養液分別進行了高效液相色譜檢測。檢測結果發現,在28 ℃,120 r/m的培養條件下,培養24 h時,野生型T23培養液中檢測不到膠毒素,在48、72、96 h的培養液中均檢測到膠毒素;而在相同的培養條件下,基因敲除突變體ΔgliN-T在4 個生長時期的培養液中均未檢測到膠毒素(表3)。

(a) 薄層板在254 nm紫外光下的薄層檢測結果;(b) 經過蒸汽碘顯色后的薄層色譜圖 1和5:膠毒素標準品;2和4:野生型T23培養液萃取樣品;3:突變菌株培養液萃取物。 (a) chromatography under 254 nm UV light; (b) chromatography after vapor iodine staining 1 and 5, gliotoxin standard substrate; 2 and 4, extraction from wild type T23; 3, extraction fromΔgliN-T.
圖4 薄層色譜法檢測T23及突變體膠毒素
Fig.4 Gliotoxin detection by TLC for T23 andΔgliN-T
表3 高效液相色譜檢測不同時間段培養液中的膠毒素含量
Table 3 Gliotoxin detection for different time-period culture filtrate by HPLC mAU·S
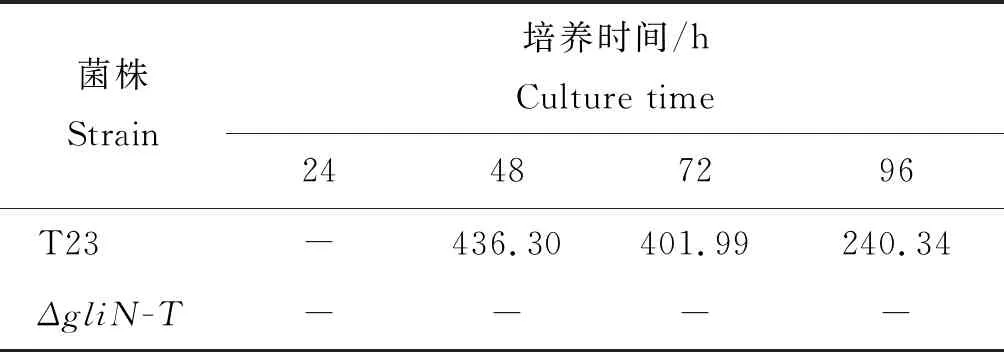
菌株Strain培養時間/hCulture time24487296T23-436.30 401.99240.34ΔgliN-T----
注:“-”表示在相同液相色譜條件下未檢測到峰值。
Note: - represents no peak is detected under the same HLPC condition.
3 討論與結論
高效率的基因敲除技術是進行絲狀真菌基因功能研究的重要生物技術之一。目前應用于絲狀真菌基因敲除的轉化技術包括分生孢子電擊轉化[20]、基于菌絲原生質體的PEG/CaCl2轉化[21]、基因槍轉化及農桿菌介導的遺傳轉化(ATMT)[22]等。其中,ATMT技術因成本低、操作簡單、轉化效率高、遺傳穩定等優點已經被廣泛應用到各類絲狀真菌的遺傳轉化中[23]。農桿菌介導的遺傳轉化技術雖然已經在綠木霉菌(T.virens)中有成功應用的報道[24-25],但是轉化效率低,大大限制了T.virens重要基因的功能驗證及開發利用。本研究對ATMT技術進行改良,在T.virensT23的分生孢子液中添加細胞壁溶解酶,轉化效率是對照組的40 倍,并通過改良的ATMT技術獲得膠毒素合成候選基因gliN-T的基因敲除突變體,這將為深入研究T.virensT23膠毒素合成的分子調控機制提供了強有力的技術支撐。
甲基轉移酶是一類普遍存在于生物體內的重要酶類,參與生物體的甲基化反應,對生物體次生代謝產物的合成具有重要的調控作用[26-27]。在煙曲霉膠毒素合成基因簇內存在著2 個甲基轉移酶基因(gliN及gliM),但是,尚未見有關煙曲霉膠毒素合成基因簇內甲基轉移酶基因的功能研究報道。
本研究利用改良的ATMT技術對gliN-T進行基因敲除并獲得突變體,發現敲除gliN-T基因后,T.virensT23膠毒素的合成受到抑制,gliN-T參與了調控T23膠毒素的合成。在膠毒素合成基因簇內存在著gliN-T及gliM-T2 個甲基轉移酶基因,這2 個基因甲基化底物是否相同?其在調控膠毒素合成途徑中各扮演著什么角色?這些問題有待深入探索。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
