
聚陰離子型鈉離子電池正極材料的研究進展
2020-06-18 11:52:18潘雯麗關文浩姜銀珠
物理化學學報 2020年5期
關鍵詞:結構
潘雯麗,關文浩,姜銀珠
浙江大學材料科學與工程學院,硅材料國家重點實驗室,杭州 310027
1 引言
近年來,風能、太陽能等清潔能源發展迅猛,發電量不斷上升,但這些間歇式能源在并入電網時易引發頻率不穩定的問題。為了確保電網的平穩運行,需要實時保持發電量與負荷之間的平衡,而電池儲能為電網的高效調峰調頻提供了可能1。綜合考慮電池的成本與儲量兩大因素,室溫鈉離子電池成為發展大規模固定式儲能系統最具競爭力的電池之一2,3。與鋰離子電池類似,鈉離子電池主要由正極、負極、隔膜、電解液和集流體組成,其充放電過程一方面依靠鈉離子在正負極之間嵌入脫出從而在內部形成通路,另一方面通過電子在電極上得失并傳輸得以在外電路產生電流。在這個過程中,正極材料往往提供電池工作時需要的鈉離子,且在很大程度上決定著電池所能提供的輸出電壓,是電池的重要組成部分,也是鈉離子電池的研究重點。
典型的鈉離子電池正極材料分為過渡金屬氧化物,聚陰離子化合物和普魯士藍類似物三類4-8。其中,聚陰離子化合物由聚陰離子基團和過渡金屬元素組成,由于聚陰離子對材料的氧化還原電對具有可調的誘導效應,使得人們容易實現對這類材料電位的提升從而獲得高電位的正極材料。另外,聚陰離子化合物的結構較為穩定,有利于實現長期循環,熱穩定性普遍較好,具有較高的安全性,這些優點使其在大規模固定式儲能系統方面有較為廣闊的前景5。
從材料應用角度來說,作為平衡電網電力供應和需求水平的儲能器件,鈉離子電池需要快速存儲和輸出能量,即要求電極材料可以快速傳導離子和電子。聚陰離子型正極材料通常可提供含有豐富離子擴散通道的儲鈉框架,離子在體相內的擴散取決于通道的維度和對載荷離子的作用。然而,在聚陰離子化合物結構框架中,過渡金屬離子往往被不傳導電子的聚陰離子基團分隔,其價電子的電子云因孤立而阻礙了電子交換,致使材料的本征電子電導率極低,限制了聚陰離子正極的實際應用9,10。因此,提高電子電導率就成為改善聚陰離子正極材料倍率性能的關鍵。此外,在材料的科學研究領域,對電極材料本征的充、放電機理的深入研究是評估及改進材料性能的關鍵,若電極材料的動力學性能不足以支撐電極反應正常發生,則會掩蓋材料的充、放電性能,給材料的反應機理探索帶來巨大困難。因此,足夠好的動力學性能也是電極材料反應機制研究的基礎。
本文主要從材料結構和材料修飾兩個方面,對決定聚陰離子正極材料電荷傳輸能力的本征影響因素和提高整體電極材料倍率性能的策略進行闡述和總結,為發展高倍率的聚陰離子型正極材料提供基本理論和實踐依據,也可作為其它高倍率鈉電正極材料開發的參考。
2 鈉離子電池的工作原理與電荷傳輸
鈉離子電池具有與鋰離子電池相似的工作原理和儲能機制,電池結構與工作原理示意圖如圖1所示11。在充、放電過程中,鈉離子電池依靠鈉離子在正、負極之間可逆的穿梭實現電荷的輸運,進而引起電勢的變化,實現電能的儲存和釋放,是一種典型的“搖椅式”電池。充電時,鈉離子受到一定的驅動力,從正極材料的結構中脫出并遷移到對電極。這一過程的驅動力由濃度梯度和電場梯度共同提供,二者合稱電化學勢。電化學勢決定著離子遷移的方向,但是僅憑電化學勢分析離子在正極材料內的傳輸過程是遠遠不夠的。從微觀上看,一定溫度下,離子在平衡位置隨機跳躍,而在一定的驅動力作用下將偏離平衡位置,形成宏觀上的凈擴散現象。在正極材料中,鈉離子通常占據間隙位,并通過空位進行傳輸。間隙位的大小和間隙位周圍的化學環境都會對擴散的速率和路徑產生影響,更準確地分析通常需要借助第一性原理進行計算,例如通過對橄欖石結構的LiFePO4進行計算,發現其(010)方向是鋰離子擴散勢壘最低的方向12。離子從晶格中脫出時,由于電中性的要求,過渡金屬離子的價態將升高,這一氧化反應的涉及到電子轉移,需要電極材料具有一定的電子電導率。因此,對于正極材料,若要實現高倍率充、放電,既須要結構框架內的鈉離子能夠在通道中順暢地擴散并在電極界面實現嵌入和脫出,也要求電子能快速地通過電極材料內部和界面傳輸到外電路中。而只要離子或電子中的一種在正極活性材料中傳導不順利,就會限制整體電極材料的倍率性能發揮,乃至影響鈉離子電池整體的功率輸出。
電池的電極反應在電極/電解質界面上發生,鈉離子需要從主體材料中擴散到界面參與電極反應或者經界面遷移至主體材料中,這一過程發生的難易程度鈉離子在材料內的擴散系數以及擴散距離有關,其擴散的特征時間可由公式表示為:
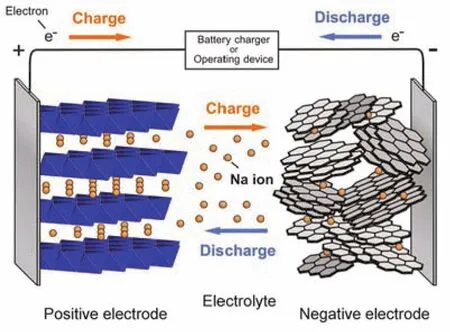
圖1 鈉離子電池的工作原理示意圖11Fig. 1 The working principle of SIBs 11.

其中Lion是鈉離子的擴散距離,與活性材料的顆粒尺寸有關,DNa為鈉離子在主體材料中的擴散系數,由材料的結構決定,為本征特性13。對于聚陰離子化合物,過渡金屬與氧原子組成的多面體往往被陰離子與氧原子組成的多面體分隔,金屬的電子云處于不連續的狀態,參與電化學反應的電子很難在電極材料里快速傳輸,因而電子傳輸往往成為聚陰離子型材料參與電化學反應時的約束步驟,大大限制了其倍率性能發揮。綜合電極反應發生涉及到的兩種載流子的傳輸特點,可以看出,本征特性決定了正極材料倍率性能的下限,而對整體電極材料形態的設計和改進可以提高其倍率性能的上限。因此,在設計和開發高倍率電極材料時,不僅須要密切關注電極材料整體動力學性能的改進策略,更應深刻了解活性材料本征的離子擴散系數和電子傳導率及其影響因素。
3 常見聚陰離子型正極材料的結構和鈉離子擴散系數
“結構決定性能”是材料科學中一條顛撲不破的真理,欲了解電極材料的本征性質和反應機理,進而設計高倍率正極材料,對于材料結構本身的把握是最基礎的。在鈉離子電池正極材料中,大量的工作對材料的結構進行了設計和探究從而調控鈉離子在其中的擴散。鈉離子的擴散系數與主體材料的結構有關:多維離子通道有利于鈉離子向多個方向遷移;而離子通道尺寸越大、路徑越平直,結構框架對鈉離子擴散的阻礙越小,鈉離子就更容易在通道中進行直接擴散。下面對不同聚陰離子型的鈉離子電池正極材料進行介紹,來說明不同結構對鈉離子擴散系數的影響。
3.1 正磷酸鹽
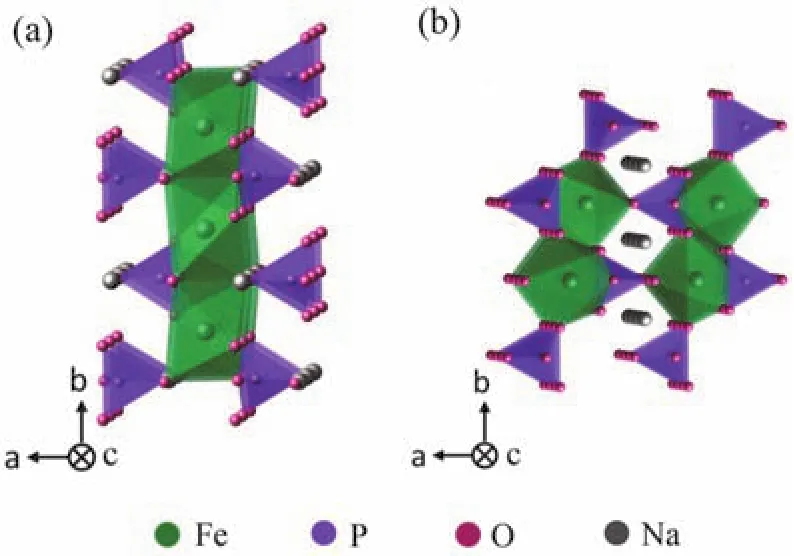
圖2 NaFePO4的結構示意圖Fig. 2 The crystal structures of NaFePO4.
對于鋰電中的磷酸鹽材料,以磷酸鐵鋰(LiFePO4)為例,其具有適中的電位(~3.6 VvsLi/Li+),低廉的成本和長循環壽命,目前已經得到了廣泛的應用14-16。磷酸鐵鋰的成功使得人們在研究鈉離子電池時,自然把目光投向了同為正磷酸鹽的磷酸鐵鈉(NaFePO4)。NaFePO4有兩種結構,一種為olivine相,一種為maricite相,兩者的晶體結構如圖2所示17-19。二者都為正交晶系,空間群為Pnma,但原子在其中的排列方式不同。Olivine相和maricite相中磷原子均與四個氧原子形成PO4四面體,鐵原子與六個氧原子形成FeO6八面體。不同的是,前者的FeO6八面體以共點形式連接,平行于bc面形成層狀結構,層與層之間由PO4四面體連接起來,構成沿c軸的一維鈉離子通道,因而具有電化學活性,但是因其為熱力學非穩定相,不能直接合成,只能通過電化學在FePO4嵌鈉的方法得到;而后者結構中FeO6以共邊形式連接,沿b軸形成一維鏈狀,再由PO4四面體連接形成三維的框架結構,這種結構中并沒有鈉離子的傳輸通道,因而通常認為maricite不具有電化學活性,但Kim等20于2015年發現通過降低顆粒尺寸以及非晶化轉變,不僅縮短了離子擴散距離,還在結構中制造了隨機的離子擴散路徑,使得maricite中所有的鈉離子都能夠在結構中可逆地嵌入脫出,提供145 mAh·g-1的容量和2 V左右的平均電位,說明結構中是否有離子擴散路徑決定了材料能否發揮出電化學性能。
3.2 焦磷酸鹽
由于磷的電負性不夠強,導致磷酸根僅能帶來較弱的誘導效應,maricite相和olivine相的NaFePO4的平均電位均低于2.5 V,Barpanda等21,22開發了陰離子基團具有更高電負性的焦磷酸根鐵基電極材料—Na2FeP2O7。Na2FeP2O7由FeO6八面體和PO4四面體組成,其中FeO6共點連接形成Fe2O11的二聚物,再通過P2O7以共點形式連接構成三維框架,在[100]、[10]和[01]三個方向都具有鈉離子傳輸的通道。Na2FeP2O7通過Fe2+/Fe3+提供3 V的平均電位,在沒有任何優化時即可在C/20倍率下提供82 mAh·g-1的容量,10C倍率下仍具有接近50 mAh·g-1的容量,表明材料具有良好的離子傳輸特性和適中的電子電導率。這項研究為高倍率正極材料的開發提供了一個方向—多維通道結構材料。此外,同為焦磷酸鹽的Na2CoP2O7也受到了關注。Kim等23通過調控缺陷,獲得了不同結構的Na2CoP2O7,進而改變了鈉離子在其中的擴散路徑。Na2FeP2O7和Na2CoP2O7的結構如圖3所示。
3.3 鈉快離子導體(NASICON)
另一類被廣泛研究的磷酸鹽是以Na3V2(PO4)3為代表的NASICON結構材料24,25。Na3V2(PO4)3基于V3+/V4+氧化還原對,可以提供120 mAh·g-1的比容量以及3.4 V的電位平臺,且具有優異的離子傳輸特性,成為最具優勢的鈉電正極材料之一。而它突出的離子傳輸特性與其獨特的NASICON結構是分不開的。如圖4a所示,Na3V2(PO4)3由獨特的“燈籠式”骨架單元組成,每個單元包含兩個VO6八面體和三個PO4四面體,并共點連接,形成[V2(PO4)3]結構基元,“燈籠式”結構之間通過PO4四面體連接。Na3V2(PO4)3提供容納鈉離子的兩類離子占據位,其中,2個鈉離子有序占據18e位置,可從主體材料中脫嵌,形成NaV(PO4)326。NASICON這種高度開放的框架結構為鈉離子提供了三維擴散通道和很大的遷移間隙,因此,鈉離子在這類材料中的擴散系數十分引人關注,可達10-10cm2s-127。由于具有NASICON結構的材料在倍率性能方面有著十分突出的優勢,人們在Na3V2(PO4)3的基礎上大量研究了具有相似結構的電極材料,例如其他金屬部分取代釩,改變電極電位,開發出Na3VMn(PO4)3、Na3VFe(PO4)3、Na3VNi(PO4)3、Na3VZr(PO4)328,29;引入氟增強陰離子基團的電負性,獲得具有更高電位的Na3V2(PO4)2F330,利用釩氧易結合的特點得到基于V4+/V5+的Na3(VO)2(PO4)2F331,最近,鄭州大學的陳衛華副教授和伍倫貢大學的侴術雷研究員開發出低成本的NASICON正極材料Na4Fe3(PO2)(P2O7),具有優良的儲鈉性能,在20C倍率下具有69.1%的容量保持率32。這些材料并未改變NASICON結構框架,因此均表現出了優異的倍率性能。可見,對于具有高離子擴散系數的結構,在改善其他電化學性能時,為了不犧牲倍率性能,應以不破壞材料結構框架作為先決條件。
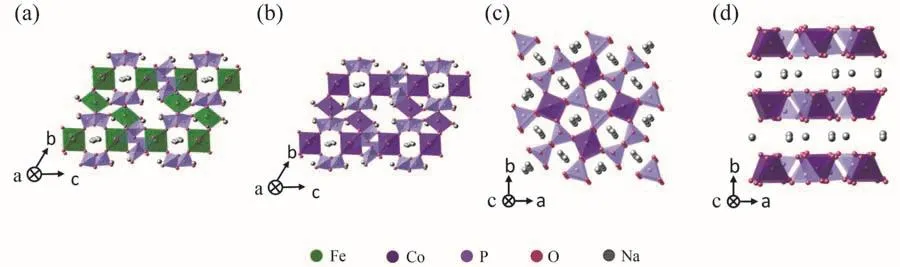
圖3 焦磷酸鹽鈉電正極材料的結構Fig. 3 The structures of pyrophosphate cathodes for SIBs.
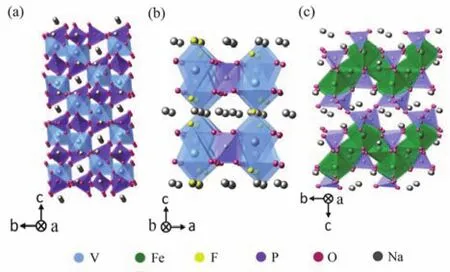
圖4 NASICON型磷酸鹽鈉電正極材料的結構Fig. 4 The structures of NASICON-type phosphate cathodes for SIBs.
3.4 其他材料
其它陰離子基團的材料,如硅酸鹽有Na2FeSiO433-35,Na2MnSiO436-38,Na2CoSiO439,40,硫 酸 鹽 有 Na2Fe(SO4)2·2H2O41-44,Na2Fe(SO4)2·4H2O45, NaFeSO446-48和Na2+2xFe2-x(SO4)3(x= 0-0.4)49-52等等,結構如圖5和圖6所示,這些材料具有不同的結構和鈉離子傳輸的特性。常見的聚陰離子化合物的結構和對應的鈉離子擴散系數表1所示。值得一提的是,本文作者所在課題組對硅酸鐵鈉結構進行了研究,發現硅酸鐵鈉結構中雖沒有擴散通道,但仍具有可觀的離子擴散系數。原因在于硅酸鐵鈉中鐵與氧為少見的四配位關系,FeO4四面體和SiO4四面體交替連接形成穩固的框架,而鈉則為六配位,單獨形成亞晶格,由于結構框架中離子間隙大,鈉離子運動自由度大,鈉離子在結構中處于相對無序的狀態,可實現自由擴散。因此,在探索和設計高倍率正極材料時,對于材料結構應辨證看待,以多維離子擴散通道為綱,但不應把擴散通道作為唯一標準,也因綜合分析特定材料的特點。

圖5 硅酸鹽鈉電正極材料的結構Fig. 5 The structures of orthosilicate cathodes for SIBs.
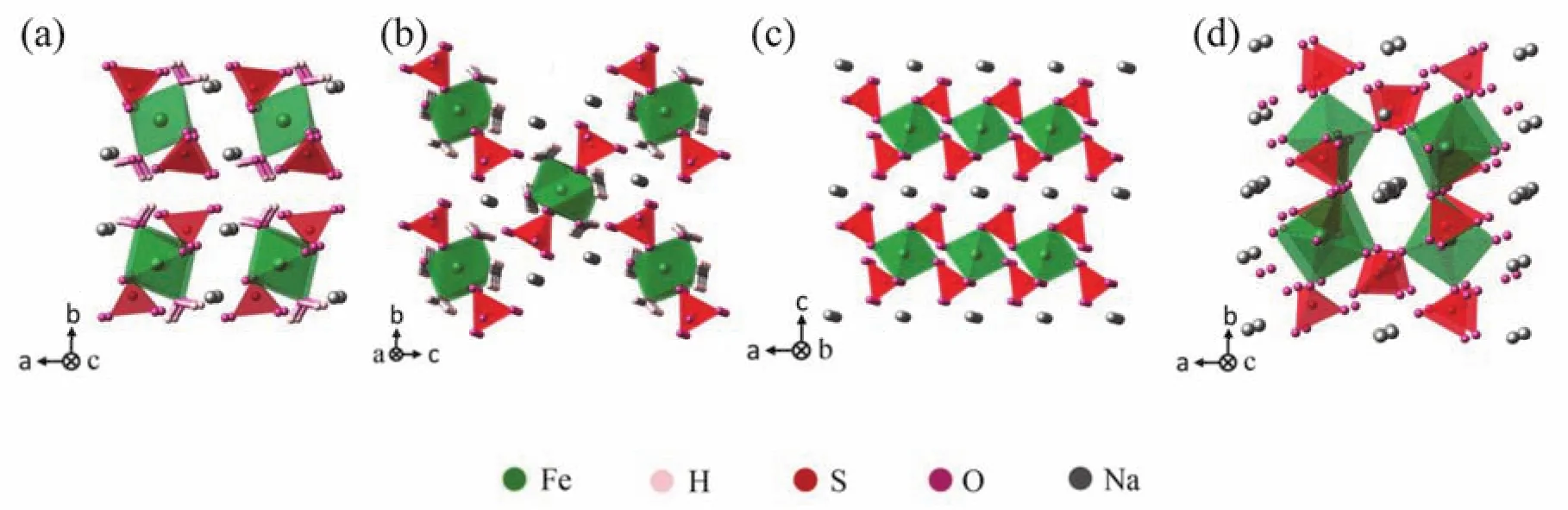
圖6 硫酸鹽鈉電正極材料的結構Fig. 6 The structures of sulfate cathodes for SIBs.
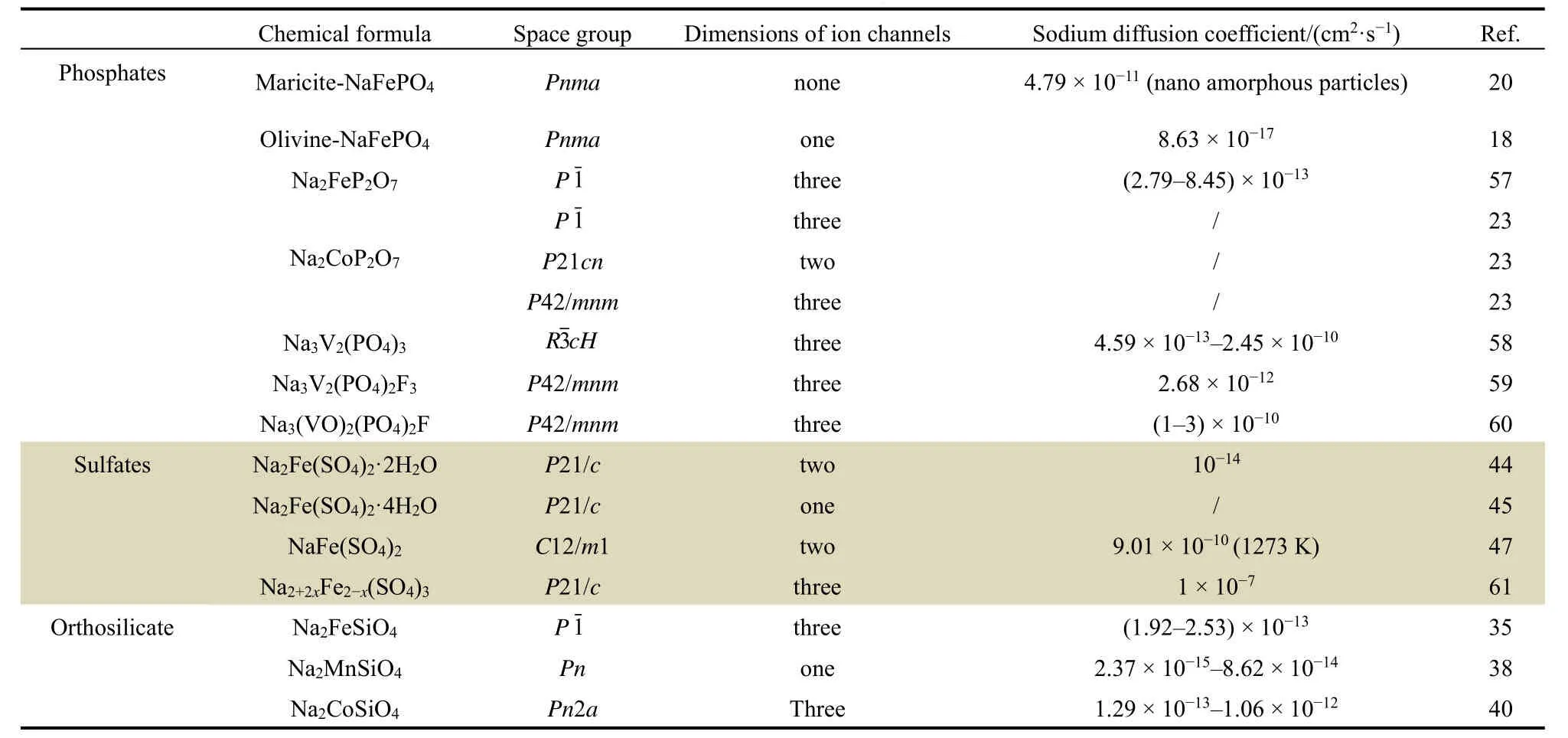
表1 不同聚陰離子鈉電正極材料的結構及離子擴散系數Table 1 Structures and sodium ion diffusion coefficients of polyanion cathodes for sodium ion batteries.
上文提到的鈉離子的擴散系數可以通過實驗進行測定,常用的方法有循環伏安法(CV)、電化學阻抗譜(EIS)和恒電位間歇滴定(PITT)等53-56。需要注意的是,這幾種測量方法測定的是表觀的擴散系數,可能受電極材料的表面狀態影響較大,僅能在一定程度上反映鈉離子在材料中擴散的難易程度。
4 提高倍率性能的策略
除了鈉離子擴散系數等材料固有的性質,影響倍率性能的因素還有鈉離子的擴散距離以及電極材料的電子電導,二者均涉及整體電極材料的界、表面問題和較為宏觀的材料形態設計問題,也是目前研究最多、最全面的方面。針對這兩個問題,提高電極材料倍率性能的方法有表面修飾,調控形貌,以及納米化等。采用上述方法的一種或多種,材料的倍率性能通常能夠得到有效提高。常見聚陰離子鈉電正極材料的倍率性能如表2所示。
4.1 表面修飾
活性材料顆粒表面是電極反應發生的場所,反應發生的難易程度與電極材料表面形態和穩定性直接相關,因此,修飾表面是改善電極材料動力學性能最直接、有效的策略,也是材料工程應用領域頗為關注的課題。參考鋰電正極材料磷酸鐵鋰改性方法,在鈉電聚陰離子型正極材料的表面工程中,碳修飾同樣是最常見的提高倍率性能的手段。碳材料容易獲取,導電性良好,包覆在聚陰離子化合物顆粒的外層不僅可以構成優良的導電網絡,還可以分散活性材料顆粒并限制其長大,有效地增大活性材料的比表面積,縮短鈉離子擴散距離,促進離子傳輸。同時,碳材料較耐電解液腐蝕,可減少電解液與活性材料的副反應,并可以作為顆粒間的緩沖層減小充放電過程中材料的體積膨脹帶來的影響,提高材料的循環壽命。基于以上優點,多種形態的碳材料被用于與聚陰離子化合物材料復合,最常見的是將有機物與材料前驅體混合并通過熱分解在產物顆粒表面原位包覆碳層,作者所在課題組也采用這種方法對幾乎電子絕緣的硅酸鐵鈉和硫酸鐵鈉材料進行了表面修飾35,71。我們利用廉價的蔗糖或抗壞血酸作為碳源,碳源在350 °C下熱分解,殘留的碳包覆在電極材料顆粒表面。電化學性能測試證明,硅酸鐵鈉與硫酸鐵鈉正極只有在包覆碳后才能表現出較為全面的性能特點,且與純材料相比,碳包覆材料的倍率性能也得到了大幅提升。這種方法簡便,成本低廉,但由于殘留碳多為無定形狀態,石墨化困難,因此電子導電性還有很大的提升空間,而且受制備條件影響大,容易包覆不均勻。針對這些問題,很多工作采用了直接將導電性更好的碳,如納米球(零維),碳納米管(一維),石墨烯或其他納米碳層(二維)等,分散到前驅體中進行高溫燒結的方法,同樣也獲得了一定成效72。Longoni等57將Na2FeP2O7與多壁碳納米管復合,在1C和10C倍率下比容量分別為86和68 mAh·g-1,實現了優異的倍率性能。Jung等73結合溶膠凝膠法與固相法將Na3V2(PO4)3與氧化石墨烯(GO)簡單復合,在0.2C倍率下即可展現出90.6 mAh·g-1的容量。盡管與未加入GO的Na3V2(PO4)3相似,復合后的Na3V2(PO4)3顆粒依然團聚明顯,但在30C下依然可以提供43.2 mAh·g-1的容量,而未復合的Na3V2(PO4)3在10C倍率下幾乎無法提供容量。可見在電極材料中引入碳構筑完整導電網絡促進電子的傳導,是提升材料的倍率性能非常簡單且行之有效的方法。但這種非原位復合的碳無法對電極材料顆粒的生長進行限制,容易造成碳和活性材料顆粒各自局部富集團聚,非活性的碳材料通常用量大,碳顆粒也無法對活性材料進行保護,對電極材料的能量密度和循環性能有損害。值得注意的是,由于復合的碳材料本身不具有電化學活性,碳的引入會增加電池里非活性材料的質量,一味地通過提高碳包覆地比例來提升活性材料的電化學性能可能反而使得電極材料整體輸出的能量減少,因此碳材料應盡可能薄而均勻地包覆在活性材料周圍,以實現電池能量密度的最大化。作者認為,將上述原位碳包覆與非原位碳復合兩種手段結合起來,即將碳包覆顆粒與少量導電碳進行復合,或可達到既構筑完整導電網絡提高電極動力學性能,又可保護電極改善循環性能的目的。
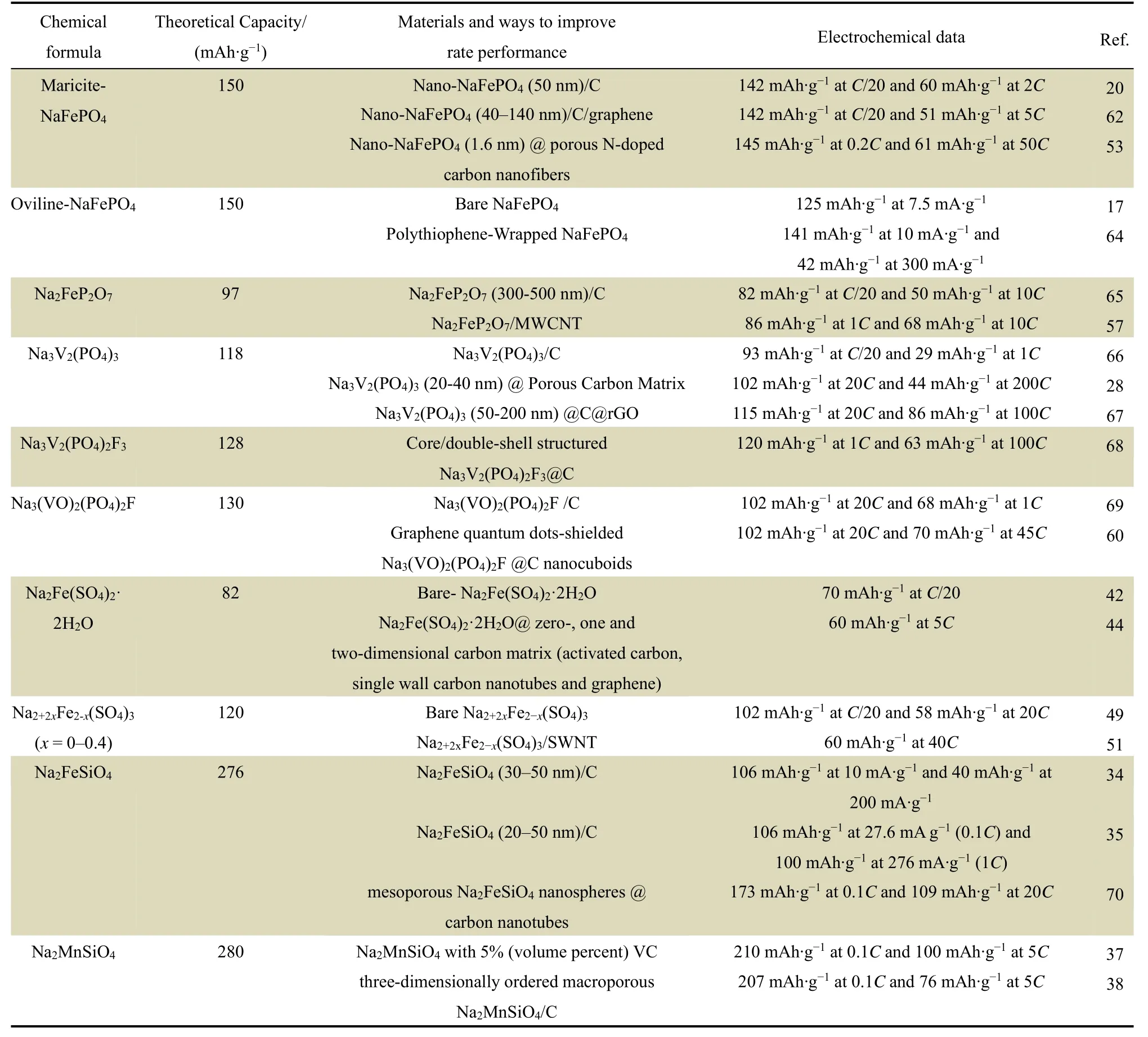
表2 不同聚陰離子鈉電正極材料的倍率性能Table 2 Rate performance of polyanion cathodes for sodium ion batteries.
除了碳材料的表面修飾,鈉離子電池正極材料表面還可以用導電高分子進行包覆,如聚吡咯(PPy)、聚噻吩(PTh)以及聚噻吩的衍生物PEDOT等等。前人報道有用PTh包覆olivine NaFePO4以及PEDOT包覆Na3V2(PO4)3,都取得了明顯的成效64,74。導電高分子的包覆條件比較溫和,通常在水溶液進行,不用后期的熱處理,但電導率通常不及碳材料,因此多用于熱穩定性相對較差的電極材料的改性。
4.2 納米工程
研究電極材料的納米結構是鈉離子電池領域又一重要課題,如上文提及,它涉及材料的形態對電極界、表面、活性位點以及鈉離子傳輸距離的影響,同時也涉及材料電子導電網絡的構建問題,這些使得電極材料的形貌調控成為另一種常用的改善動力學性能的策略。從公式(1)可以看出,由于冪次的不同,減小離子的擴散距離比提高擴散系數更為有效,這一方法也更具有普適性,從這一點看,活性材料的顆粒應盡量小以最小化離子擴散距離。正極材料的納米化可簡單理解為,通過構筑多孔、片狀形貌或者不同維度的納米結構,如納米線、納米棒等來增大材料的比表面積,進一步促進電子和離子在界面的傳輸速率。由于納米顆粒比表面積大,表面能提高,容易出現團聚現象。目前,材料納米結構的調控通常依靠活性材料外層的碳材料來實現,但與上文表面修飾部分不同的是,納米化是將顆粒至少在一個維度上限制在納米尺度,而不僅僅是簡單的復合。Li等75采用溶液輔助固相法將尺寸約5 nm的Na3V2(PO4)3顆粒分散分別分散在乙炔炭黑(AC)、碳納米管和石墨烯上,其中Na3V2(PO4)3與乙炔炭黑復合的形貌如圖7a,b所示,在5C下能獲得100.6 mAh·g-1的比容量,并且循環200圈后的容量保持率為96.4%。隨后,他們還將Na3V2(PO4)3與還原氧化石墨烯(rGO)復合,成功制備了層層交疊的結構76。rGO層不僅促進了電子的傳輸,還限制了Na3V2(PO4)3只能在rGO層間以納米片狀形式長大,形貌如圖7c,d所示。這種復合結構在5C倍率下的比容量可以達到111 mAh·g-1,500次循環后仍具有102 mAh·g-1的比容量。Shen等60報道了石墨烯量子點復合的Na3(VO)2(PO4)2F納米棒,表面形貌如圖7e,f所示。Na3(VO)2(PO4)2F納米棒沿[001]方向擇優生長,極大的縮短了鈉離子在ab晶面的傳輸距離,石墨烯量子點在表面形成一層電子傳輸網絡,并且減少了Na3(VO)2(PO4)2F納米棒的團聚現象。Na3(VO)2(PO4)2F@GO納米棒的構筑極大地提高了離子和電子的傳輸速率,在45C倍率下仍具有70 mAh·g-1的比容量,2000次循環后容量保持率超過80%。
由以上敘述可見,正極材料的納米化從形態學角度對鈉離子在材料中的擴散予以解釋,可對材料的動力學性能提升提供最直觀的理解和幫助。但材料的納米結構越多樣化,雖然可能對倍率性能有很直接有效的幫助,但也意味著對電極材料的體積評估難度越大,甚至會犧牲材料應用層面非常關注的體積能量密度,因此,我們在設計正極材料的納米結構時,應綜合評估材料納米化對能量密度、動力學性能和穩定性,避免矯枉過正。
4.3 分級結構
上文所述的納米顆粒可以縮短離子遷移的距離,進而提升反應的動力學,但由于比表面積大,表面能高,難以在導電劑和粘結劑中分散,導致較高的接觸阻抗,并且在充放電過程中,由于離子的遷移、擴散,納米顆粒可能會發生電化學團聚現象,致使納米化的效果降低。為了既保持納米顆粒的高倍率性能,同時解決納米化后帶來的一系列問題,電極材料的分級結構設計成為了一個有效的手段。
微-納分級結構,即微米尺度的二次顆粒由納米尺度的一次顆粒組成,可以協同納米及微米結構的優勢。An等77設計了一種由Na3V2(PO4)3/C納米片自組裝形成微米花的分級結構,如圖8a,b所示,直徑為1-2 μm的微米花由20-40 nm厚的納米片組成。這種微-納結構既增大了材料與電解液接觸的面積,縮短了離子傳輸路徑,又成功抑制了納米片的電化學團聚,在1 A·g-1(9.1C)的電流密度下具有86 mAh·g-1的比容量,5000次循環后容量保持率為83.6%。Fang等78從碳框架的角度對電極材料進行了分級結構設計,利用CVD在Na3V2(PO4)3顆粒外層包覆一層碳,在顆粒間形成碳納米纖維構成導電網絡,結構如圖8c,d所示。這種分級碳框架的復合材料既促進了電子在單個材料顆粒的傳輸,又提高了顆粒間的電子傳輸速率,在500C的超大倍率下仍具有38 mAh·g-1的比容量,在30C倍率下循環20000次后容量保持率為54%。
分級結構的設計有利于材料在循環過程中保持納米結構,進而保持高倍率的性能。值得注意的是,制備分級結構的材料需要對工藝參數有較好的把握,對于不同的活性材料,可能需要改變碳源才能達到最佳的效果。
4.4 元素摻雜和取代

圖8 (a,b)分級結構的Na3V2(PO4)3/C微米花的掃描圖像77;分級結構的碳框架與Na3V2(PO4)3復合的(c)掃描圖像及(d)透射圖像78Fig. 8 (a, b) TEM images of hierarchical Na3V2(PO4)3/C microflowers 77; (c) SEM and (d)TEM images of hierarchical carbon framework wrapped Na3V2(PO4)3 78.
在上文中,我們提到,聚陰離子化合物的結構決定了其極低的電子電導率,屬于材料本征性質的范疇。因此,為了提升電子電導率促進材料結構層面的電荷傳輸,我們將關注點轉向更深層次的材料結構上。
可作為鈉離子電池正極材料的聚陰離子化合物的結構由三個要素組成:堿金屬離子、過渡金屬離子和聚陰離子基團。由于鈉離子主要在擴散通道中傳輸,而這部分只涉及結構框架中的電荷交換,因此不對堿金屬離子進行討論。正極材料在充放電過程中,電荷交換總是伴隨著結構中過渡金屬離子的氧化還原,因此電子的傳導路徑依賴于過渡金屬-氧多面體之間的相互連接。但聚陰離子化合物中存在電子絕緣的聚陰離子基團,它們往往與過渡金屬-氧多面體鍵接,阻隔了電子傳輸路徑。因此,欲提升材料的本征電子電導率,必須在結構框架中創造額外的電荷轉移途徑。主要有兩種方法,其一是通過額外金屬離子的引入,其中同價金屬離子創造金屬多面體鍵接點;異價離子則通過電荷補償機制提升電子電導率,但這方面的工作目前并沒有系統報導。另外一個方案是進行非金屬位的異價取代,這種方法可以讓陰離子-氧鍵斷裂,異價取代機制也在陰離子基團中引入電荷交換途徑,聚陰離子基團也能傳導電子,從而提高材料的本征電導率,但目前這方面的研究工作較少。作者所在課題組利用陰離子位的異價取代的方法,成功加速了硅酸鐵鈉正極材料結構中的電荷交換,改性后的硅酸鐵鈉材料在552 mA·g-1的大電流密度下仍可以保持大于200 mAh·g-1的比容量,而且循環穩定,目前這項工作的研究還在繼續。
5 總結與展望
高倍率、低成本的鈉離子電池在儲能領域有著廣闊的應用前景,而作為電池中鈉離子的供給來源,正極材料的動力學性能將直接影響電池的倍率性能。本文選擇了穩定性和安全性出色的聚陰離子型正極材料作為討論對象,首先探討了決定材料動力學性能的本征因素—材料的結構,在選擇與設計材料結構時,應以多維擴散通道為目標,但低維或不存在擴散通道的材料也可通過不同的方法創造擴散路徑以提高動力學性能。而后本文綜述了目前聚陰離子型正極材料動力學性能的改進策略,如通過表面修飾、納米工程和分級結構的設計從電極材料整體形態的角度提高倍率性能,或通過元素摻雜或取代的方法提高材料本征的電導率。目前對于本征性質的分析還不夠,在未來的工作中,我們應更多從材料本身的角度出發,為鈉離子的擴散機理或添加劑的作用機制提供更本質、更科學的解釋。由于鈉離子電池與鋰離子電池具有相似的工作原理,一些用于提升鋰離子電池電極性能的手段相對更加成熟,也值得借鑒79-83。另外,在選擇優化策略時,應具體材料具體分析,針對材料的特點進行特異性優化,避免盲目選擇改進方案。
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
