β-伴大豆球蛋白α-亞基核心區的基因克隆、表達及純化
2020-07-03 07:10:46李垚熹袁艷秋高夢楠周樹義胡亞云欒廣忠
食品與機械 2020年5期
李垚熹 袁艷秋 巨 倩 高夢楠 周樹義 胡亞云 欒廣忠
(西北農林科技大學食品學院,陜西 楊凌 712100)
大豆貯藏蛋白的主要成分為β-Conglycinin(7S大豆球蛋白),是一種由α′、α、β3種不同亞基構成的三聚體[1-2],占大豆蛋白的30%,其對大豆蛋白的結構功能特性影響顯著。研究[3-5]表明,α′及α亞基的結構可分為核心區(core region)和延展區(extension region),且均含有兩個N-連接糖基;β-亞基則不具備延展區,只有一個N-連接糖基。Maruyama等[6-7]研究發現,在N-連接糖基及延展區缺失后,原核宿主的核心區仍能自組裝成三聚體。阻礙7S大豆球蛋白凝集的原因可能在于其糖基具有較強的親水性[8-9],因此提高7S大豆球蛋白的凝膠能力的關鍵或許在于去除糖基。目前,重組蛋白純化領域中起重要作用的是親和標簽,其中最普遍使用的是His標簽。許妍妍等[10-11]采用基因克隆技術、基因重組技術以及蛋白分離純化技術成功制備了缺失N-連接糖基的重組α’-亞基。試驗擬選取品種為齊黃34的大豆種子提取大豆總RNA,使用大腸桿菌作為原核宿主對β-伴大豆球蛋白的α-亞基核心區進行克隆與表達,并利用AKTA蛋白純化系統通過鎳離子親和層析柱對其進一步純化,為后期重組大豆蛋白分散體系的構建提供依據。
1 材料與方法
1.1 原料、載體與菌種
大豆:齊黃34,山東省農科院;
感受態細胞E.coliDH 5α、E.coliBL21(DE3)、質粒pET-28a:西安熱默爾生物科技有限公司;
質粒pGEM-T Easy:普洛麥格(北京)生物技術有限公司。
1.2 試劑與儀器
MiniBEST Plant RNA Extraction Kit、PrimeScriptTMII 1st Strand cDNA Synthesis Kit、DNA純化試劑盒、質粒提取試劑盒、蛋白質分子量 Marker(3597Q)、rTaq DNA聚合酶、DL 2000 DNA Marker、DL 5000 DNA Marker、Quick Cut限制性內切酶EcoR Ⅰ、Xho Ⅰ:寶生物工程(大連)有限公司;
T4-DNA連接酶:普洛麥格(北京)生物技術有限公司;
瓊脂粉、β-巰基乙醇、異丙基硫代-β-D-半乳糖苷(IPTG)、丙烯酰胺、十二烷基硫酸鈉(SDS):北京索萊寶有限公司;
其余試劑為國產分析純;
PCR擴增儀:Hema 9700型,珠海黑馬醫學儀器有限公司;
超微量紫外—可見分光光度計:NanoPro 2010型,北京鼎昊源科技有限公司;
冷凍離心機:Neofuge 15R型,上海力申科學儀器有限公司;
通風櫥:CAV1000型,肯尼亞F&S Scientific公司;
超聲波細胞破碎儀:VC130型,美國 Sonics & Materials公司;
蛋白純化系統:ClearFirst-3000型,上海閃譜生物科技有限公司。
1.3 試驗方法
1.3.1 引物的設計與合成 在NCBI(https://www.ncbi.nlm.nih.gov/)中查找7S大豆球蛋白α-亞基的基因序列,GB編號為NM_001249927.2(Glycine max beta-conglycinin alpha-subunit 〔CG-3〕, mRNA),依據編碼核心區的基因序列設計一對特異性引物,即上游引物F1:5′-CCGACGACGACGACAAGGATTCTGAGTTACGAAGA-3′,下游引物R1:5′-CCGCTCGAGTTCAGTAAAAAGCCC-3′。考慮到蛋白純化后N-端標簽的處理,設計上游引物時引入了腸激酶酶切位點(加粗部分)及其保護堿基;為方便后續克隆,在下游引物中引入了內切酶Xho Ⅰ酶切位點(加粗部分)、終止密碼子(斜體部分)及相應的保護堿基,理論上擴增所得的基因片段大小為1 300 bp左右,引物合成由寶生物工程(大連)有限公司完成。
1.3.2 目的基因的擴增 將齊黃34大豆種子使用液氮研磨成粉末,取300 mg凍干粉提取總RNA,而后以此為模板運用試劑盒自帶的Random 6 mers構建cDNA單鏈。具體操作步驟:42 ℃逆轉錄1 h,70 ℃保溫15 min,4 ℃恒溫保存。以cDNA單鏈為模板利用自行設計的引物F1/R1對目的基因αc進行擴增,選用10 mg/mL的瓊脂糖凝膠電泳對目的基因進行檢測并回收,PCR反應體系參考Maruyama等[6]的方法修改如下:PrimeSTAR Max Premix(2×) 25 μL,F1(20 μmol/L) 0.5 μL,R1 (20 μmol/L) 0.5 μL,反轉錄反應液4 μL,無菌純水補加至50 μL。PCR反應條件為98 ℃變性10 s,66 ℃退火5 s,72 ℃延伸30 s,35個循環,最后72 ℃延伸7 min。
1.3.3 重組克隆載體的構建、篩選及鑒定 參照許妍妍等[10-11]的方法并修改。通過T4-DNA連接酶將目的基因片段αc與選用的克隆載體pGEM-T Easy相連接,將連接體系于16 ℃反應1 h,導入感受態細胞E.coliDH 5α中。在含有氨芐青霉素的LB固體培養基中隨機挑取4~5個獨立的白色菌落用于菌落PCR篩選。菌落PCR反應體系:聚合酶rTaq mix 26 μL,單克隆菌株菌液2 μL,F1/M47 (20 μmol/L) 2 μL,F2/M48 (20 μmol/L) 2 μL,添加去離子水至50 μL。使用氨芐青霉素濃度為100 μg/mL的液體LB培養基對篩選出的陽性克隆菌株進行培養14 h,提取質粒,選用10 mg/mL的瓊脂糖凝膠電泳及XhoⅠ/EcoRⅠ雙酶切對所提質粒進行鑒定。首先通過XhoⅠ內切酶酶切重組質粒5 min,再加入EcoRⅠ內切酶繼續酶切15 min。將鑒定結果與預期相符的質粒進行外送測序(由西安熱默爾生物科技有限公司完成),將測序結果與NCBI的α-亞基核心區的基因編碼區進行序列比對[12],將測序符合預期的構建成功的重組克隆載體命名為pGEM-αc,將陽性克隆菌株于37 ℃進行增菌培養后,于-80 ℃保存菌種。
1.3.4 重組表達載體的構建、篩選及鑒定 使用限制性內切酶XhoⅠ/EcoRⅠ對重組克隆載體pGEM-αc和選用的表達載體pET-28a進行雙酶切,酶切反應體系:待酶切載體10 μL,內切酶XhoⅠ/EcoRⅠ各1 μL,10× Quick Cut Green Buffer緩沖液5 μL,添加去離子水至50 μL。將二者的雙酶切體系于37 ℃反應15 min后回收產物,通過10 mg/mL的瓊脂糖凝膠電泳對產物進行鑒定并回收具有雙黏性末端的目的片段。
將上述兩種目的片段回收后,在T4-DNA連接酶作用下于16 ℃連接1 h,連接體系(20 μL):pGEM-αc雙酶切產物5 μL,pET-28a雙酶切產物10 μL,50% PEG 2 μL,T4-DNA連接酶1 μL,T4-DNA連接酶10×Buffer 2 μL。將連接成功的產物導入到感受態細胞E.coliBL21(DE3)中,選用卡那霉素濃度為50 μg/mL的固體LB培養基對轉化菌液進行培養,同時設置陽性與空白對照,37 ℃培養14 h。隨機挑取若干獨立的單克隆菌落,利用特異性上游引物F1和通用下游引物T7er對其進行菌落PCR篩選。將篩選出的陽性菌株接種于卡那霉素濃度為50 μg/mL的液體LB培養基中培養14 h,提取質粒,使用10 mg/mL瓊脂糖凝膠電泳及Xho Ⅰ/EcoR Ⅰ單、雙內切酶酶切對質粒進行鑒定,將鑒定結果與預期相符的質粒外送測序。以pET-28a-αc為測序結果正確的重組表達載體,以pET-28a-αc-BL21為陽性菌株,挑選單菌落的陽性菌株于37 ℃下進行增菌培養,菌種置于-80 ℃保存[13-14]。
1.3.5 重組蛋白的提取 將pET-28a-αc-BL21的菌液培養6 h,以體積分數為1%的接種量接種至含有卡那霉素50 μg/mL的液體LB培養基中,繼續培養至菌液濃度OD600 nm值為0.8時,加入0.2 mmol/L的誘導劑IPTG進行誘導表達,于37 ℃持續培養9 h,取1 mL誘導菌液用于SDS-PAGE電泳檢測(分離膠體積分數12%,濃縮膠體積分數5%),通過Quantity One軟件分析目的蛋白表達量[15-17]。其余菌液于4 ℃,5 000 r/min離心15 min,收集菌體,使用磷酸鹽緩沖液PBS (25 mmol/L磷酸鈉緩沖液,500 mmol/L NaCl,1 mmol/L EDTA,1 mmol/L PMSF,0.02% NaN3,pH 7.4)對菌體重懸后通過冰浴超聲破碎菌體[8],超聲條件:振幅25,6 s/4 s,30 min。超聲4~5個循環后取出,取1 mL全菌液用于后續SDS-PAGE分析,其余破碎菌體于4 ℃,8 500 r/min離心45 min,收集上清液即為目的蛋白α-亞基核心區的粗蛋白液,于4 ℃保存,將收集的全菌液、上清液和沉淀進行SDS-PAGE分析[18-19]。
1.3.6 重組蛋白α-亞基核心區的純化 使用AKTA蛋白純化系統對重組α-亞基核心區的粗蛋白液進行純化,將制備好的100 mL粗蛋白溶液通過孔徑為0.45 μm的水性濾膜過濾后,于4 ℃儲藏,使用自動上樣泵上樣到預先用磷酸鹽緩沖液平衡好的His Trap HP預裝柱上,先使用5~10倍柱體積的磷酸鹽緩沖液洗脫未與鎳離子柱結合的雜蛋白,于280 nm下檢測并收集各蛋白峰直至峰值達到基線,使用20%,50%,80%,100%的含有500 mmol/L咪唑的磷酸鹽緩沖液對樣品進行洗脫并收集對應的蛋白峰。將收集到的所有蛋白峰通過SDS-PAGE電泳進行分析鑒定[20-22]。
1.3.7 數據處理 使用NCBI官網的Blast (http://blast.ncbi.nlm.nih.gov/Blast.cgi)功能對測序結果進行對比分析;使用UniProt官網(http://www.uniprot.org/blast/)查找7S大豆球蛋白α-亞基核心區的氨基酸序列并使用Blast功能進行比對;使用DNA MAN軟件分析重組表達載體的堿基序列及編碼氨基酸;使用軟件Snap Gene分析重組表達載體的測序結果;使用軟件Quntity One分析目的蛋白α-亞基核心區的表達量;使用Image Lab分析核酸電泳凝膠圖。
2 結果與分析
2.1 大豆總RNA的提取
由圖1可知,1、3泳道的兩個條帶較為清晰,分別為28S和18S,證明提取的RNA并未被大量降解;而2、4泳道中的兩個條帶稍顯模糊,尤其是2泳道中的條帶彌散比較明顯,說明所提取的RNA可能存在大部分被降解的情況,因此使用超微量紫外—可見光分光光度計進行進一步測定。1~4泳道的RNA樣品濃度分別為96.65,35.22,85.94,53.95 ng/μL,各樣品的A260 nm/A280 nm分別為2.04,1.75,1.97,1.95。綜上,2、4泳道中提取的RNA樣品質量不高,因此選取1、3泳道提取的大豆總RNA用于后續的RT-PCR試驗。
2.2 目的基因的擴增
由圖2可知,所得基因片段位置為1 000~1 500 bp,長度約為1 300 bp,與α-亞基核心區基因的理論值(1 265 bp)一致,證明α-亞基核心區基因片段擴增成功。

M. DL 5000 DNA Maker 1~4. 大豆總RNA

M. DL 5000 DNA Maker 1. α-亞基核心區基因片段
Figure 2 Amplification of target geneα-subunit core region by RT-PCR
2.3 重組克隆載體的構建、篩選及鑒定
由圖3可知,采用自行設計的特異性引物F1/R1對隨機選取的5個菌株擴增得到的片段長度均為1 300 bp左右,與目的基因的理論值相一致;而使用通用引物M47/M48擴增得到的片段長度為2 000 bp左右,是因為特異性引物與通用引物的位點中間有一定的片段大小,因此該結果也與理論值相符。理論上而言,重組克隆載體經單酶切后應形成清晰而單一的條帶,但pGEM-αc經EcoRⅠ單酶切后形成兩個清晰條帶,其大小分別與克隆載體和目的基因的理論值相符,是因為選用的克隆載體pGEM-T Easy上帶有EcoRⅠ酶切位點;重組克隆載體經Xho Ⅰ單酶切后能形成單一條帶,證實重組克隆載體構建成功;而經雙酶切后重組克隆載體被切成兩個較為清晰的條帶,且大小分別為3 000 bp與1 300 bp左右,與載體pGEM-T Easy(3 015 bp)和目的基因α-亞基核心區的理論值(1 265 bp)相符。
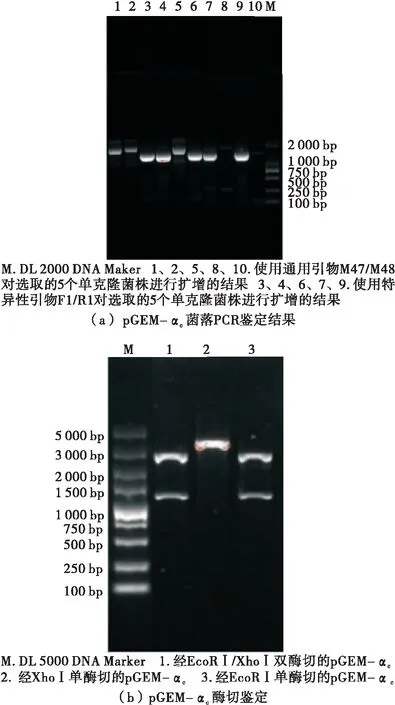
圖3 重組克隆載體pGEM-αc的鑒定及篩選
Figure 3 Identification and screening of recombinant cloning vector pGEM-αc
2.4 重組表達載體的構建、篩選及鑒定
由圖4可知,4、5、6、12泳道擴增所得產物條帶大小為1 000~2 000 bp,約為1 300 bp,與目的基因理論長度相符,而其余泳道未出現目的條帶,可能是因為挑選的菌落為假陽性。考慮到條帶的清晰度、彌散程度和亮度,選用4、12泳道進行后續試驗。pET-28a-αc被限制性核酸內切酶XhoⅠ、EcoRⅠ分別單酶切后均可得到與理論值大小相符的單一條帶;而被限制性核酸內切酶XhoⅠ、EcoRⅠ雙酶切后則得到兩個條帶,片段大小分別為1 300,5 000 bp左右,與α-亞基核心區(1 265 bp)和載體pET-28a(5 369 bp)的大小相符。通過測序比對可知,該序列與UniProt中編號為P0DO15的氨基酸序列相似度達到100%,未出現堿基移碼和錯配的現象,證明重組表達載體構建成功。
2.5 重組蛋白的存在形式
由圖5可知,未添加誘導劑的工程菌pET-28a-αc-BL21經超聲破碎后,其全菌液、上清液和沉淀中(1~3泳道)均未在50 kU附近出現目的蛋白條帶;而37 ℃下誘導培養的菌體經超聲后的全菌液、上清液和沉淀中(4~6泳道)均于50 kU處出現目的蛋白條帶,由于上清液中所含的重組蛋白未形成包涵體,具有較好的溶解性,可收集用于進一步的分離純化[13,23];蛋白純化的穿透峰及洗脫峰濃縮后的濾液(8泳道)、誘導培養的菌體超聲后的上清液經蛋白純化的穿透峰及其濃縮液(9~10泳道)中無目的蛋白條帶,與期望相符的,證明在使用低濃度咪唑洗脫雜蛋白時并沒有目的蛋白被洗脫下來;而添加誘導劑的菌體超聲后的上清液經蛋白純化的洗脫峰及其濃縮液(11~14泳道)中有目的蛋白的單一條帶,證明洗脫峰中目的蛋白含量較高,且目的蛋白含量為80%左右。
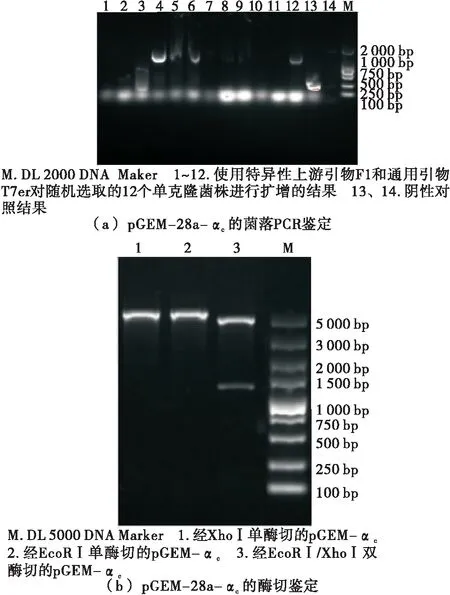
圖4 重組表達載體pGEM-28a-αc的鑒定及篩選
Figure 4 Identification and screening of recombinant cloning vector pGEM-28a-αc
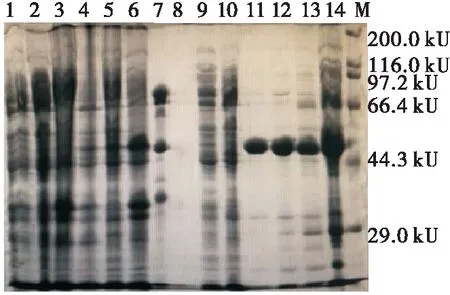
M. 蛋白質Marker 1~3. 未誘導工程菌的全菌、上清液和沉淀4~6. 誘導的全菌、上清液和沉淀 7. 天然7S 8. 蛋白純化峰濃縮后的濾液 9~10. 蛋白純化的穿透峰及濃縮液 11、13. 蛋白純化洗脫峰 12、14. 蛋白純化洗脫峰的濃縮液
圖5 重組α-亞基核心區的提取及純化電泳圖
Figure 5 Electrophoresis of the extraction and purification of the recombinantα-subunit core region
3 結論
從齊黃34品種的大豆種子中提取總RNA,通過自行設計的特異性引物F1/R1經RT-PCR對7S大豆球蛋白α-亞基核心區的基因進行擴增,成功構建了經Xho Ⅰ/EcoR Ⅰ雙酶切鑒定、菌落PCR鑒定以及堿基測序均正確重組克隆載體pGEM-αc和重組表達載體pET-28a-αc。將重組表達載體pET-28a-αc導入到感受態細胞E.coliBL21(DE3)中,經IPTG誘導表達獲得分子量約為50 kU的重組α-亞基核心區蛋白,其表達條件為:OD600 nm值為0.8、IPTG濃度為0.2 mmol/L、誘導溫度為37 ℃、誘導時間為9 h,此條件下重組α-亞基核心區蛋白可以得到大量表達,其純度為87%以上。如何提高α-亞基核心區蛋白制備效率及純度還需進一步研究。
