高含固率木質纖維素厭氧發酵物總DNA的4種提取方法比較
2020-07-28 09:09:46馬旭光劉素君常佳麗
中國沼氣 2020年2期
馬旭光,劉素君,江 滔,常佳麗,唐 瓊
(樂山師范學院 化學學院,四川 樂山 614000)
厭氧發酵技術因能將農作物秸稈和禽畜糞便等農業纖維質固體廢棄物轉化為清潔能源生物天然氣(經提純后使甲烷含量>95%的沼氣)而備受關注,同時對于推動我國循環農業的發展具有重要意義[1-2]。厭氧發酵產甲烷的本質是由水解、酸化、產乙酸細菌和產甲烷古菌等多種微生物協同作用將復雜的大分子有機物逐步分解轉化為甲烷和二氧化碳的過程[3]。高含固率(發酵體系中固含物>10%)厭氧發酵技術較傳統的低含固率發酵技術具有容積產氣率高、沼液排放量小、能耗低等優點,目前已成為糞秸高效產甲烷領域的研究熱點[4-5]。
認識厭氧發酵體系中功能性微生物群的構成、生長代謝規律及其生態生理學以達到定向調控厭氧發酵過程是該領域重要的研究方向之一[6-8]。近年來隨著分子生物學技術的快速發展,尤其是宏基因組技術在揭示生物多樣性領域中的廣泛應用,使得從DNA水平對不可培養的微生物種類進行全面研究成為可能[9]。能否獲得高質量的厭氧發酵物中微生物總DNA直接關系到分子技術分析結果的客觀性和準確性。
目前從土壤、腸道、堆肥、發酵床墊料、厭氧顆粒污泥、沼液等環境樣本中提取微生物總DNA的方法主要有十二烷基磺酸鈉(sodium dodecyl sulfate, SDS)法,十六烷基三甲基溴化銨(cetyltrimethyl ammonium bromide, CTAB)法,SDS-CTAB結合法以及生物技術公司提供的試劑盒法[10-15]。筆者前期采用上述幾種方法提取高含固率(>10%)的牛糞和玉米秸稈混合厭氧發酵物中微生物總DNA,質量均不高,導致在后續聚合酶鏈反應(polymerase chain reaction, PCR)中無法獲得理想目的條帶。究其原因,可能與高含固率木質纖維素物料在厭氧發酵過程中產生的大量腐殖酸、酚類、糖類以及雜蛋白等不利于PCR擴增的物質有關[16]。另外,高含固率糞秸厭氧發酵物較廢水等液體厭氧發酵物具有質地不均一、微生物濃度低、雜質多等特點[17],而且在厭氧發酵體系中木質纖維素底物主要靠吸附在其上的細菌通過分泌胞內酶來完成分解[18],這些因素均會影響提取微生物總DNA的質量。但目前還見專一性提取高含固率纖維質厭氧發酵物中微生物總DNA方法的報道。
基于此,本研究針對該類樣本的特點,在前人研究基礎上,對傳統的SDS-CATB法進行了改進,提取不同高含固率糞秸厭氧發酵物的微生物總DNA,并與常規的SDS法,SDS-CATB法和淤泥試劑盒法進行比較,通過定性分析DNA電泳條帶、特異片段PCR擴增效果和定量分析DNA的純度和濃度,評價該方法的有效性和穩定性,以期針對高含固率纖維質厭氧發酵物建立一種高效、低成本的微生物總DNA提取方法。
1 材料與方法
1.1 樣本與前處理
樣本來源:提取總DNA的樣本為4種中溫(37℃±1℃)批次厭氧發酵6 d的纖維質混合物:玉米秸稈和牛糞、油菜秸稈和牛糞、油菜秸稈和豬糞、油菜秸稈和雞糞,均取自實驗室正常產甲烷的批次厭氧反應器,含固率在12.3%~14.5%,其理化性質見表1。
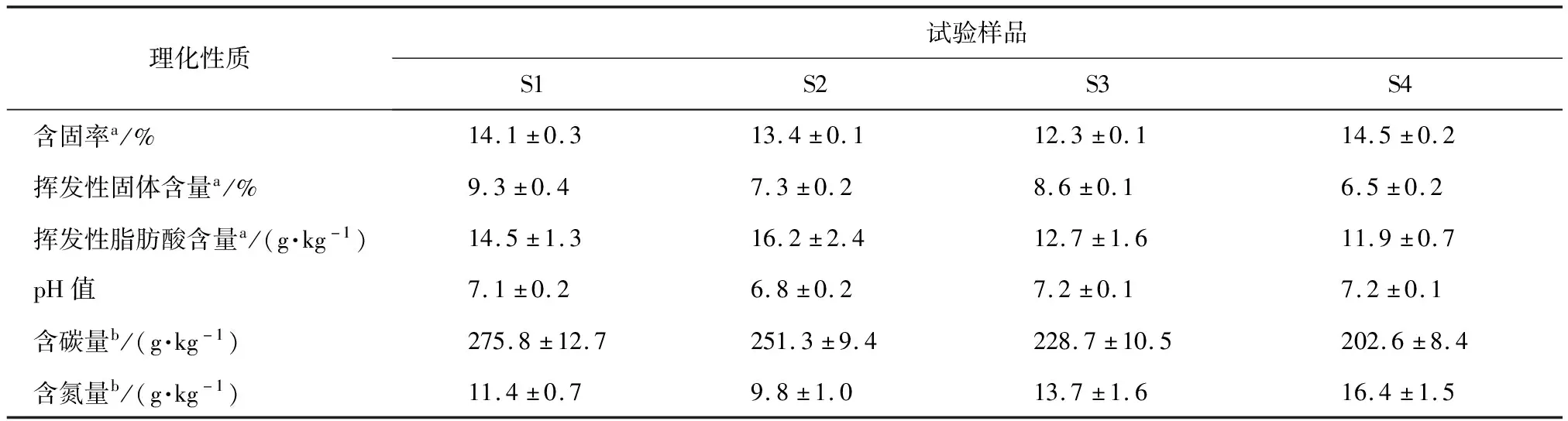
表1 試驗樣品的理化性質
樣本前處理方法:稱取2 g左右(準確記錄重量)上述糞秸發酵物于無菌的10 mL離心管中,5000 rpm冷凍離心10 min,棄上清液后往沉淀加入3~5倍TE buffer(pH值8.0)緩沖液,混勻后-40℃備用。
1.2 試驗內容
先以玉米秸稈和牛糞混合發酵物(S1)為試驗樣本,分別用試劑盒法,SDS法,SDS-CTAB法和改進的SDS-CTAB法提取微生物總DNA,比較提取DNA的質量和PCR擴增效果,每個處理設3~4個重復;然后為了進一步驗證改進SDS-CTAB法的有效性和穩定性,分別取1 g左右油菜秸稈和牛糞(S2)、油菜秸稈和豬糞(S3)、油菜秸稈和雞糞(S3)的混合厭氧發酵物提取微生物總DNA進行復證研究,每個處理設3個重復。
1.2.1 試劑盒法
采用北京百泰克生物技術有限公司生產的糞便總DNA提取試劑盒(離心柱型),具體操作方法見說明書。
1.2.2 SDS法
參照趙勇[19]等提取土壤微生物總DNA的方法。
1.2.3 SDS-CTAB法
參照劉云浩[13]等提取微生物發酵床墊料中微生物總DNA的方法。
1.2.4 改進的SDS-CATB法
1.2.4.1 樣品表面微生物的洗脫方法
取上述-40℃保存的樣品于室溫融化后,加入2~3倍于提取物體積的林格氏液,渦旋混勻,5000 rpm離心10 min,棄上清液。反復以上步驟2~3次,離心的沉淀物用于下一步雜質的去除。
1.2.4.2 腐殖酸和雜蛋白質的去除方法
將1.2.4.1中獲得的沉淀物中加入2~3倍于提取物體積的洗滌液,渦旋混勻,5000 rpm離心10 min,棄上清液。反復以上步驟,直至上清液清澈透明后,離心的沉淀物用于下一步DNA的提取。
1.2.4.3 微生物細胞壁的裂解方法
將1.2.4.2中經處理過的沉淀樣品中加入1~2倍體積的SDS裂解液,100 μL的蛋白酶K,100 μL的溶菌酶,150 μL的復合纖維素酶,200 μL的異硫氰酸胍(GuSCN)洗液,渦旋混勻后,65℃水浴45 min。
1.2.4.4 微生物總DNA的沉淀和純化方法
(1)將1.2.4.2中水浴后的樣品5000 rpm離心10 min,用移液槍轉移褐色上清液至10 mL滅菌離心管。
(2)根據轉移上清液的體積,加入0.2倍體積的3M醋酸鈉無菌溶液和1倍體積的10%聚乙二醇(polyethylene glycol, PEG)-8000無菌溶液,-20℃下靜置30 min后,5000 prm離心10 min,棄上清液,留黃色沉淀。
(3)往留有黃色沉淀的離心管中加入1倍體積的CTAB裂解液,65℃水浴10 min后,迅速加入等體積氯仿:異戊醇(24∶1),上下顛倒,輕輕混勻,5000 rpm離心10 min。
(4)將1.2.4.3中的上清液一次性轉移至無菌的5 mL離心管中,加入等體積的10%PEG-8000溶液,置于4℃冰箱中8 h,沉淀DNA。
(5)將1.2.4.4中的提取物12000 rpm 離心10 min,棄上清液后加入500 μL的70%冰乙醇洗滌沉淀后,12000 rpm 離心10 min后棄上清液。
(6)將上述步驟(5)中的沉淀物于超凈工作臺吹干后,加入100 μL無菌超純水溶解DNA,并保存于-40℃待后續分子生物學分析。
1.2.5 DNA定性檢測方法
配置1.2%的瓊脂糖凝膠,Goldview II核酸染料用量為瓊脂糖凝膠溶液體積的0.2‰,DNA的檢測量為5 μL,電泳緩沖液為1×TAE,電泳時間為20 min,100 V電壓,超純水脫色,在凝膠成像儀中于302 nm紫外線下檢測并采集電泳圖。
1.2.6 DNA純度和定量檢測方法
用紫外線分光光度計中檢測提取DNA的A260/A280,A260/A230和濃度值。
1.2.7 16S rRNA基因的PCR擴增方法
提取DNA的細菌16S rRNA基因在PCR擴增儀中完成,引物為357F-GC和517R,由北京六合通經貿公司合成。引物357F-GC的序列為GC clamp-5’-CCTA CCTACGGGAGGCAGCAG-3’,其中GC-clamp序列為5’-CGCCCGCCGCGCGCGGCGGGCGG GGCGGGGGCACGGGGGG-3’,引物517R的序列為5’-ATTACCGCGG CTGCTGG-3’[20]。擴增反應體系為50 μL(見表2),DNA marker分子量為2000 bp,模板DNA的濃度為10 ng·μL-1(可根據提取DNA的濃度進行適當倍數稀釋)。擴增程序為:95℃預變性10 min,25個熱循環(93℃變性1 min,48℃退火1 min,72℃延伸1 min 30 s),最后72℃延伸5 min[21]。
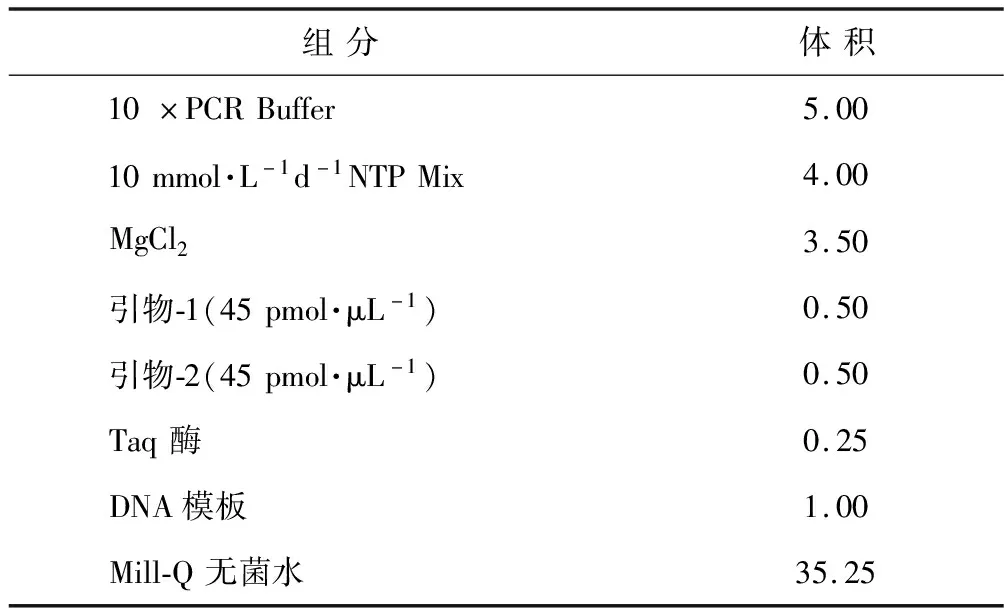
表2 PCR反應體系(50 μL)的組成 (μL)
1.3 主要試劑和儀器
1.3.1 試驗試劑
(1)林格氏液:NaCl 9 g,KCl 0.12 g,CaCl20.24 g, NaHCO30.2 g,充分溶解后,定容至1L[22]。
(2)TE buffer(pH值8.0):取5 mL 1 mol·L-1的Tris-HCl Buffer(pH值8.0)和1 mL 0.5 mol·L-1EDTA(乙二胺四乙酸)(pH值8.0)用超純水定容至500 mL。
(3)洗滌液(pH值10):取50 mL 1mol·L-1Tris-HCl,40 mL 0.5 mol·L-1EDTA,5.844 g NaCl和10 g PVP-40(聚乙酸吡絡烷酮),用超純水溶解、定容至1 L。
(4)SDS裂解液:取50 mL 1mol·L-1Tris-HCl,20 mL 0.5 mol·L-1EDTA,7.305 g NaCl,20 g SDS,10 g PVP-40,用超純水溶解,調pH值至8.0,定容至500 mL。
(5)GuSCN洗液(pH值6.4):取10 mL 1mol·L-1Tris-HCl,59.08 g GuSCN,溶解調pH值至6.4,定容至100 mL。
(6)3 mol·L-1醋酸鈉(NaOAc)溶液(pH值5.0):取123.045 g NaOAc溶于450 mL去離子水后,加25 mL 冰醋酸,充分攪拌并定容至500 mL。
(7)CTAB裂解液:取50 mL 1 mol·L-1Tris-HCl,20 mL 0.5 mol·L-1EDTA,40.908 g NaCl,10 g CTAB,10 g PVP-40,充分溶解后定容至500 mL。
(8)10%PEG-8000溶液:取50 g PEG-8000,70.128 g NaCl,用超純水溶解、定容至1 L。
上述配置的試劑均需在121℃滅菌15 min,室溫保存。所用的分子生物學等級試劑Tris base,Na2EDTA·H2O,PVP-40,SDS,GuSCN和PEG-8000以及蛋白酶K(產品貨號:V900887)均購自Sigma-Aldrich西格瑪奧德里奇(上海)貿易有限公司,溶菌酶(CAS:9001-63-2)和復合纖維素酶(CAS:9012-54-8)定制自深圳市思美泉生物科技有限公司,其余試劑均為國產分析純等級。
1.3.2 試驗儀器
高速冷凍離心機(JIDI-16R,廣州吉迪儀器有限公司),超微量紫外分光光度計(MD2000D,Biofuture, Made in Britain),電泳儀(DYY-8C,北京六一生物科技有限公司),凝膠成像儀(新JY04S-3C,北京君意東方電泳設備有限公司),16S rRNA PCR擴增儀(ProFlex PCR System, Life technologies, Made in Singapore)。
2 結果與分析
2.1 不同提取方法的總DNA純度和濃度
DNA的純度和濃度是評價提取樣本中總DNA質量的重要指標。一般而言,A260/A280表示DNA中蛋白質的純度,該值越大,說明蛋白質的純度越高[12];A260/A30表示DNA中腐殖酸的含量,該值越大,說明腐殖酸污染越嚴重,對后續PCR反應越不利[23];DNA的濃度表示樣品中總DNA的得率,該值越大,說明提取的DNA得率越高,在后續分析中更能客觀反應樣品中的微生物多樣性[24-25]。不同方法提取玉米秸稈與牛糞混合厭氧發酵物中微生物總DNA的純度和濃度檢測結果見表3。

表3 不同方法提取試驗樣品中微生物總DNA的純度和濃度
由表3可知,4種方法獲得同一樣本中的總DNA純度和濃度有明顯差異。改進SDS-CATB法提取的質量最佳,A260/A280,A260/A230,濃度分別能達到1.8,1.7和114.5 ng·μL-1,均顯著(P<0.05)高于其他3種方法,說明該方法提取的總DNA中蛋白質純度最高,腐殖酸雜質最少,DNA得率也最高。一般認為,A260/280>1.7時,蛋白質較純;A260/230>1.5時,腐殖酸雜質含量不會影響后續PCR反應[24]。糞便試劑盒法提取的A260/A280和A260/A230分別僅為1.16和0.74,說明該方法不適合提取高含固纖維質厭氧發酵物微生物總DNA。
改進SDS-CTAB法能獲得高質量DNA的原因可能與采用PVP-40洗液除雜有關。有研究表明,在緩沖液中加入適量的高分子螯合物PVP能絡合提取樣本中的多酚和萜類物質,具有抗氧化作用,進而有效防止多酚氧化為醌類物質,以免提取液變成褐色而影響DNA的純度[26-27]。在本實驗提取過程中,發現未使用洗滌液的糞便試劑盒法、SDS法和SDS-CTAB法獲得的DNA有不同程度發黃,說明在破壁前采用PVP-40洗液對去除樣本中多酚、萜類和腐殖酸等雜質起到了關鍵作用。另外,改進SDS-CTAB法有較高DNA得率的原因可能與采用林格氏液洗脫和多種酶破壁充分釋放細胞中DNA有關。
2.2 不同提取方法的總DNA和PCR產物瓊脂糖凝膠電泳效果
4種方法提取的總DNA瓊脂糖凝膠電泳檢測見圖1。由圖1可知,4種方法提取樣品總DNA電泳檢測結果有明顯不同。改進SDS-CTAB法(12~15泳道)獲得總DNA的電泳條帶單一明亮、干凈齊整,無明顯DNA降解現象,而其他3種方法的條帶均存在不同程度的拖尾和彌散現象,其中試劑盒法(1~4泳道)和SDS法(5~8泳道)尤為嚴重,說明在提取該樣品總DNA時有大量DNA斷裂碎片產生,難以獲得完整的DNA。另外,糞便試劑盒法的條帶亮度明顯低于其他3中方法,這是由于試劑盒法由于受離心管容量的限制樣本承載量為0.5 g左右,僅為其他3種手提法的1/4,且使用了純化柱純化DNA,可能導致總DNA的損失[24]。DNA瓊脂糖電泳定性分析結果與表2中超微量紫外分光光度計定量分析結果相吻合。
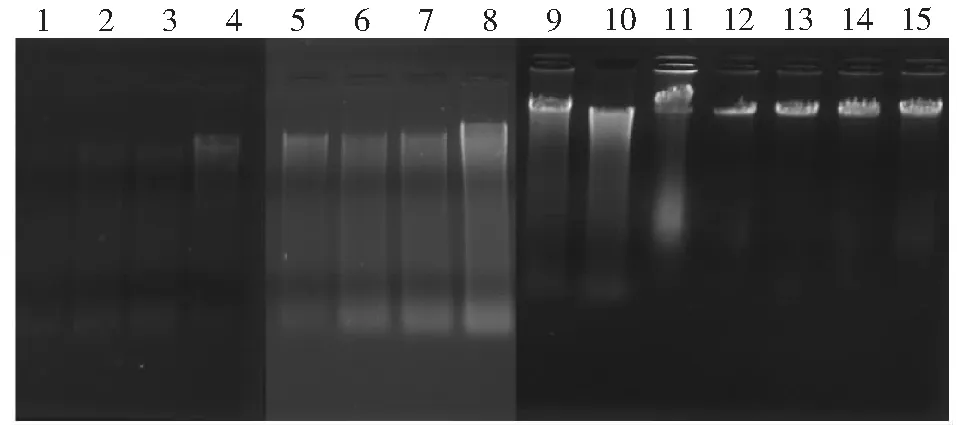
注:泳道1~4為糞便試劑盒法;5~8為SDS法;9~11為SDS-CTAB法;12~15為改進SDS-CTAB法。
4種方法提取總DNA的PCR產物凝膠電泳檢測見圖2。由圖2可知,改進SDS-CTAB法獲得的目的條帶(1~3泳道)清晰、明亮,SDS-CTAB法雖然也檢測到了擴增目的條帶(4~6泳道),但僅隱約可見,清晰度低;而SDS法(7~9泳道)和試劑盒法(10~12泳道)均未檢測到目的條帶。由于4種方法提取的總DNA濃度均能滿足PCR模板DNA適宜的濃度范圍(5~10 ng·μL-1)。由此推測,能否獲得PCR擴增目的條帶與DNA的質量密切相關。由表3可知,糞便試劑盒法和SDS法的A260/A280和A260/A230均較低,說明DNA中較低的蛋白質純度和較高的腐殖酸含量等雜質抑制了PCR反應。
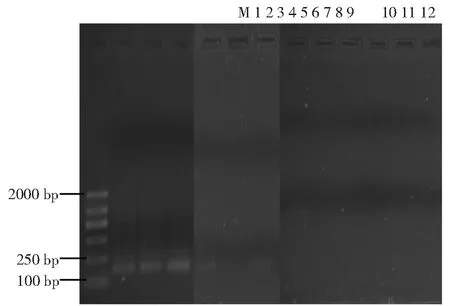
注:M為DL 2000分子量的marker;泳道1~3為改進SDS-CTAB法;4~6為SDS-CTAB法;7~9為SDS法;10~12糞便試劑盒法。
2.3 改進SDS-CTAB法提取不同樣品的總DNA純度和濃度
上述實驗結果表明,改進SDS-CTAB法提取高含固率木質纖維素厭氧發酵物料總DNA質量最高,能獲得理想的PCR擴增目的條帶,但與商業試劑盒法相比,樣本承載量較高(2 g左右),在前期洗脫和除雜階段所耗時間較長,導致日提取樣本數量有限。為此,將該方法樣本承載量降低至1 g左右,洗脫液和洗滌液添加量均提高至提取物體積的5倍,提取3種木質纖維素厭氧發酵物樣本(油菜秸稈+牛糞、油菜秸稈+豬糞、油菜秸稈+雞糞)總DNA,其純度和濃度結果見表4。
表4 改進SDS-CTAB法提取不同厭氧發酵物中微生物總DNA的純度和濃度
由表4可知,改進SDS-CTAB法提取不同樣品獲得的總DNA質量均較高,A260/A280和A260/A230均分別大于1.70和1.60,說明蛋白質純度較高,腐殖酸含量較少;總DNA的濃度均大于50 ng·μL-1,DNA得率也均較高。另外,在提取DNA過程中,發現通過減少樣本承載量,可有效縮短前期洗脫和洗滌時間,進而提高了該方法的日提取樣本數量。
2.4 改進SDS-CTAB法提取不同樣本的總DNA和PCR產物電泳效果
改進SDS-CTAB法提取3種不同樣本總DNA的瓊脂凝膠電泳和PCR產物電泳定性檢測結果見圖3。由圖3可知,不同樣本總DNA的電泳條帶均單一整齊、干凈明亮,DNA均較完整,未見明顯的斷裂和降解;獲得的PCR產物目的條帶清晰齊整,能滿足微生物多樣性的后續分析要求。由此表明,針對不同高含固率木質纖維素厭氧發酵物,采用本研究提出的改進SDS-CTAB法提取的總DNA均純度高、雜質少,性能穩定可靠,適合進一步推廣應用。
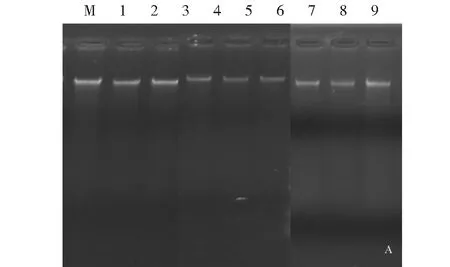
注:圖3中的1~3為油菜秸稈+牛糞,4~6為油菜秸稈+豬糞,7~9為油菜秸稈+雞糞。
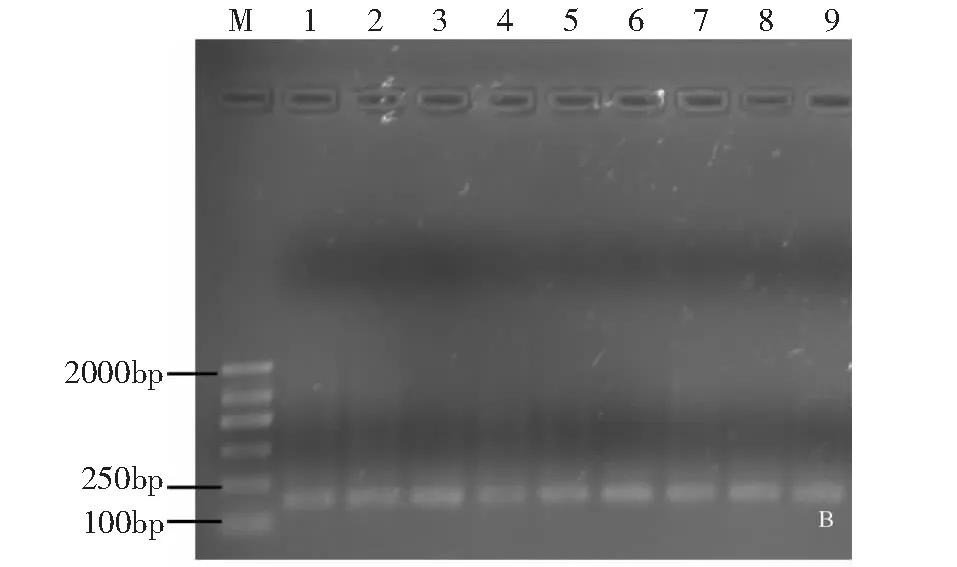
注:圖4中M為DL 2000分子量的marker;泳道編號代表的樣本與圖3相同。
3 討論
影響環境樣本微生物總DNA提取質量的因素較多,尤其是如何有效去除樣本中的殘渣、多糖、酚類、色素和腐殖酸等雜質對獲得高質量的DNA至關重要[11,23,26]。目前通常采用先富集微生物(細胞)再破壁和先破壁再離心以及化學試劑沉淀分離DNA兩種方法。Tang[23]等使用CTAB法提取糞便中微生物DNA時采用先除雜、富集微生物然后用破壁的方法,除雜效果較高且提高了DNA得率;陳競[15]等也采用類似的方法獲得了得率和質量均較高的羊糞沼液中的微生物總DNA。先破壁再純化的提取策略雖能提高DNA得率但因除雜能力有限會影響提取的質量[27],因此該方法更適用于微生物濃度較高、雜質較少的樣本。鑒于高含固率木質纖維素物料具有微生物濃度低、雜質多以及木質纖維素表面吸附有大量細菌群的特點,本研究采用先富集微生物(細胞)再破壁的提取方案,先用中性的林格氏液洗脫吸附在物料表面的微生物,以達到富集的目的,然后采用PVP-40洗滌除雜,以達到除雜的目的。研究表明,高分子螯合劑PVP能絡合樣本中的多酚、萜類物質,以及能有效防止多酚物質氧化成醌類物質而使提取變成黃色[13];高鹽溶液能降低腐殖酸和多糖對DNA的干擾[12],PEG8000也能有效減少腐殖酸和雜蛋白對DNA的污染[13]。由此可知,本研究在后續提取步驟中使用的10% PEG8000,醋酸鈉溶液和CTAB裂解液也起到了一定除雜作用。
另外,大量研究表明微生物細胞破壁方法也對DNA的得率和質量有明顯影響[13-15]。目前通常采用物理法(凍融和機械震蕩等)、化學法(SDS裂解液)和生物法(溶菌酶、蛋白酶等)裂解細胞壁,一般在實際操作中為了提高DNA得率綜合使用上述幾種破壁方法。溫洪宇[28]等的優化結果表明,凍融和SDS法聯合破壁最適合提取乳品廢水活性污泥,張琳[29]等等在提取厭氧發酵活性污泥時認為溶菌酶等生物法的破壁獲得的DNA片段長,能提高DNA得率和PCR擴增效率。鑒于本研究采用的樣品木質纖維素厭氧發酵物存在大量細胞壁較厚且致密緊實的革蘭氏陽性(G+)微生物,故采用了以生物法(裂解液、蛋白酶K、溶菌酶和復合纖維素酶)為主和化學法SDS為輔聯合裂解破壁的措施并取得理想的DNA提取效果。
綜上所述,在選用微生物DNA提取方法時不能一概而論,而應在分析提取樣本的特性基礎上,有針對性地選擇或改進現有的方法,以達到理想的提取效果。高含固率木質纖維素厭氧發酵物與廢水厭氧活性污泥、好氧發酵物以及土壤和腸道微生物等環境樣本相比有其自身特殊性,難以用上述樣本成熟的提取DNA方法。本研究針對影響DNA提取質量的關鍵步驟提出了改進SDS-CTAB法,通過林格氏液洗脫和PVP-40洗滌液除雜以及裂解液和多種生物酶聯合破壁等系列方法,獲得了質量較高的高含固率木質纖維素素厭氧發酵物總DNA。盡管手提法與商業試劑盒法相比,各提取步驟所使用的試劑成分明確且成本較低,但手提法日提取樣本量較少,不便于批量化和標準化操作。因此,今后針對本研究針對上述關鍵步驟,還需從樣本承載量、主要試劑的用量及其對微生物多樣性的影響等方面進一步優化以達到該方法標準化、商業化應用的目的。
4 結論
(1)針對不同的高含固率木質纖維素厭氧發酵物,改進SDS-CTAB法提取的微生物總DNA的A260/A280和A260/A230值能分別達到1.70和1.60以上,每克樣本的DNA濃度能達到50 ng·μL-1以上,蛋白質純度高、雜質少,穩定性好。
(2)改進SDS-CATB法的微生物總DNA電泳條帶單一齊整、清晰明亮,獲得的PCR擴增目的條帶清晰度高,適宜后續利用分子生物技術進一步分析微生物多樣性及其代謝功能。
(3)林格氏液洗脫,PVP-40洗滌液除雜以及裂解液和多種酶聯合破壁是改進SDS-CTAB法獲得高質量微生物總DNA的關鍵步驟。
猜你喜歡
兒童故事畫報(2019年5期)2019-05-26 14:26:14
豬業科學(2018年4期)2018-05-19 02:04:38
浙江農業科學(2016年11期)2016-05-04 04:16:49
Coco薇(2016年2期)2016-03-22 02:42:52
化工進展(2015年6期)2015-11-13 00:27:33
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
無機化學學報(2014年9期)2014-02-28 17:32:57
無機化學學報(2014年1期)2014-02-28 17:30:03
