禾谷鐮刀菌 Tri8基因敲除及產毒類型分析
2020-07-30 10:02:50李添夢胡小平杜光源范三紅
麥類作物學報 2020年5期
李添夢,胡小平,杜光源,范三紅
(1.西北農林科技大學生命科學學院,陜西楊凌 712100;2.西北農林科技大學理學院,陜西楊凌 712100;3.西北農林科技大學植物保護學院,陜西楊凌 712100)
赤霉病(Fusarium head blight)是影響小麥產量和品質的主要病害[1],該病害由禾谷鐮刀菌復合種(Fusariumgraminearumspecies complex,FGSC)引起,不同地域復合種的組成有所不同。引起我國小麥赤霉病的主要病原菌為禾谷鐮刀菌和亞洲鐮刀菌,其中小麥玉米輪作區以禾谷鐮刀菌為主,小麥水稻輪作區以亞洲鐮刀菌為主[2-3]。赤霉病病原菌主要侵染小麥穗部,會引起籽粒發育不良,病原菌合成的毒素在籽粒中的累積,還會對人畜健康和食品安全造成嚴重威脅[4-6]。
禾谷鐮刀菌可以合成單端孢霉烯族的真菌毒素,常見的類型有4種:脫氧雪腐鐮刀菌烯醇(deoxynivalenol,DON)、3-乙酰脫氧雪腐鐮刀菌烯醇(3-AcDON)、15-乙酰脫氧雪腐鐮刀菌烯醇(15-AcDON)和雪腐鐮刀菌烯醇(nivalend,NIV)[7-8],其中DON是帶病小麥籽粒中毒素的主要存在形式。DON毒素可以干擾核糖體肽基轉移酶的活性,阻礙核糖體的正常循環,從而抑制胞內蛋白質的合成[9]。大量攝入DON會引起急性中毒[10-11],主要表現有頭疼、頭暈、嘔吐及中樞神經系統紊亂[12]。因而,赤霉菌毒素導致的食品安全問題引起國家管理部門及研究者的高度關注。
鐮刀菌中參與毒素合成的基因有12~16個[13],命名為Tri基因,不同菌株參與的基因數目有所不同。這些基因主要集中在兩個基因簇中,其中最大的一個基因簇命名為Tri5,包含了Tri3、Tri4、Tri5、Tri6、Tri7、Tri8、Tri9、Tri10、Tri11、Tri12、Tri13和Tri14共12個Tri基因;另一個基因簇中包含了Tri1和Tri16兩個基因,Tri15和Tri101游離于上述基因簇之外[14]。DON生物合成的直接前體為法尼基焦磷酸(FPP),在Tri5基因編碼產物的催化下,法尼基首先環化形成單端孢霉二烯(TDN)[15];在Tri4編碼的P450家族單加氧酶催化下進行3次羥化反應和1次環加氧反應,形成異構單端孢霉三醇[16-17];該化合物可自發脫水環化形成異構木霉菌醇;在Tri101、Tri11和Tri3編碼產物催化下進行1次羥基化和2次乙酰化形成麗赤殼菌素(CAL);CAL在Tri1編碼的P450羥化酶催化下,在7,8兩個位置引入2個羥基[18],最終形成DON前體3,15-diAcDON;Tri8編碼分泌型酯酶,可催化3,15-diAcDON 3位或15位乙酰基的水解,從而形成3-AcDON或15-AcDON[19],不同的鐮刀菌編碼的Tri8基因有所不同,有些可催化3位乙酰基的水解,而其他則催化15位乙酰基的水解[20];3-AcDON或15-AcDON可自發,或在病菌或植物酯酶的催化下水解剩余的乙酰基,最終形成DON。
研究表明,亞洲鐮刀菌(F.asiaticum)主要產生3-AcDON和NIV類型毒素,而禾谷鐮刀菌(F.graminearum)主要產生15-AcDON[21]。其中NIV類型菌株產生的NIV毒素含量極低。本研究利用基因敲除技術獲得Tri8基因缺失的禾谷鐮刀菌株系,并對其產生毒素的類型進行了定性分析,以期為DON合成前體3,15-diAcDON的制備及Tri8蛋白體外活性分析奠定基礎。
1 材料與方法
1.1 供試材料
禾谷鐮刀菌野生型菌株PH-1和農桿菌菌株EHA105由本實驗室保存,載體pOSCAR和pA-HYG-OSCAR由本實驗室保存,PrimerSTAR DNA polymerase、PstI、BamHI、HindIII和XhoI購自Takara公司(大連),DNA快速提取試劑盒(DSBIN1151)購自廣州東盛生物科技有限公司,DNA凝膠回收試劑盒購自天根生化科技(北京)有限公司,無縫克隆試劑盒CloneExpressTM為南京諾唯贊生物科技公司。混合毒素標準品購自北納創聯生物科技有限公司(北京)。
1.2 方 法
1.2.1 基因敲除載體構建
利用CTAB法提取野生型禾谷鐮刀菌菌株PH-1基因組DNA,以其為模板,利用引物Tri8-wf-uF/uR和Tri8-wf-dF/dR(表1)分別擴增Tri8基因上下游片段Tri8-up和Tri8-down,其長度分別為1 078 bp和1 213 bp。PCR反應體系為:100 ng· μL-1的DNA模板0.5 μL、10 μmol·L-1的正反向引物各0.5 μL、2×Buffer 12.5 μL、2.5 mmol·L-1的dNTP 0.5 μL、1 U· μL-1的KOD酶0.5 μL、滅菌蒸餾水補足至25 μL。PCR 反應程序為:95 ℃預變性5 min;98 ℃變性10 s,65 ℃退火20 s,72 ℃延伸1.5 min,共35個循環;72 ℃延伸7 min。利用PstI、BamHI酶切載體pA-HYG-OSCAR,并通過膠回收獲得1 425 bp的抗性基因片段HYG。

表1 引物序列
通過重疊PCR對上述三個片段進行連接(順序為Tri8-up、HYG、Tri8-down)。PCR反應體系為:100 ng· μL-1的Tri8-up、Tri8-down和HYG基因片段各0.5 μL、2×Buffer 12.5 μL、2.5 mmol·L-1dNTP 0.5 μL、1 U· μL-1KOD酶0.5 μL、滅菌蒸餾水補足至25 μL。前期PCR反應體系為:95 ℃預變性5 min;98 ℃變性10 s,45 ℃退火20 s,72 ℃延伸3.2 min,5個循環;72 ℃延伸7 min。以上反應體系不加入引物,反應結束后加入引物Tri8-wf-uF、Tri8-wf-dR各 0.5 μL,后期PCR反應體系為;95 ℃變性5 min,98 ℃變性10 s,60 ℃退火20 s,72 ℃延伸3.2 min,35個循環;72 ℃延伸7 min。利用HindIII、XhoI對pOSCAR載體進行雙酶切,切膠回收并利用無縫克隆將其與通過重疊PCR獲得的Tri8-up-HYG-Tri8-down片段進行連接,轉化篩選最終獲得Tri8基因敲除載體pOSCAR-△Tri8。
1.2.2 農桿菌介導的轉化
利用農桿菌(EHA105)介導的轉化法將基因敲除載體pOSCAR-△Tir8轉至PH-1野生菌株,具體過程參考Zahi等[22]的報道。在陽性基因敲除菌株篩選過程中,首先利用引物HYG-F/R擴增HYG基因,初步判斷Tri8基因敲除是否成功,之后利用引物Tri8-F/R擴增Tri8基因,進一步確定Tri8基因是否成功敲除。
1.2.3 基因敲除菌株與原始菌株的產毒類性 分析
將對照菌株和Tri8基因敲除菌株接種于PDA培養基,置于25 ℃恒溫箱活化3~5 d,挑取菌塊接種至CMC培養基中,25 ℃ 200 r·min-1培養3 d。培養液用濾布過濾獲得孢子液。將孢子液加入大米培養基中,25 ℃黑暗條件下培養 30 d,取出培養基液氮速凍后研磨至粉末狀,加入400 mL甲醇,200 r·min-125 ℃過夜抽提。將培養物用三層濾紙過濾,濾液40 ℃旋轉蒸餾,濃縮至50 mL,-20 ℃保存。
2 結果與分析
2.1 基因敲除載體pOSCAR-△ Tri8的構建
Tri8基因敲除載體構建流程如圖2所示。依據禾谷鐮刀菌基因組序列設計Tri8基因上下游擴增引物,并以基因組為模版擴增獲得上下游片段Tri8-up(1 078 bp)和Tri8-down(1 213 bp)。PstI、BamHI雙酶切pA-HYG-OSCAR質粒,通過膠回收獲得潮霉素抗性基因編碼片段HYG(1 425 bp)。然后利用重疊PCR技術將三者連接獲得敲除片段Tri8-up:HYG:Tri8-down。HindIII、XhoI雙酶切pOSCAR載體,切膠回收獲得包含T-DNA左右邊界的載體,然后通過無縫克隆技術將其與敲除片斷連接,最終獲得敲除載體pOSCAR-△Tri8。利用PCR方法對敲除載體進行初步篩選,然后將陽性重組子送至楊凌天潤奧科生物科技有限公司進行測序驗證。測序結果顯示,構建的敲除載體pOSCAR-△Tri8的序列與設計完全一致。
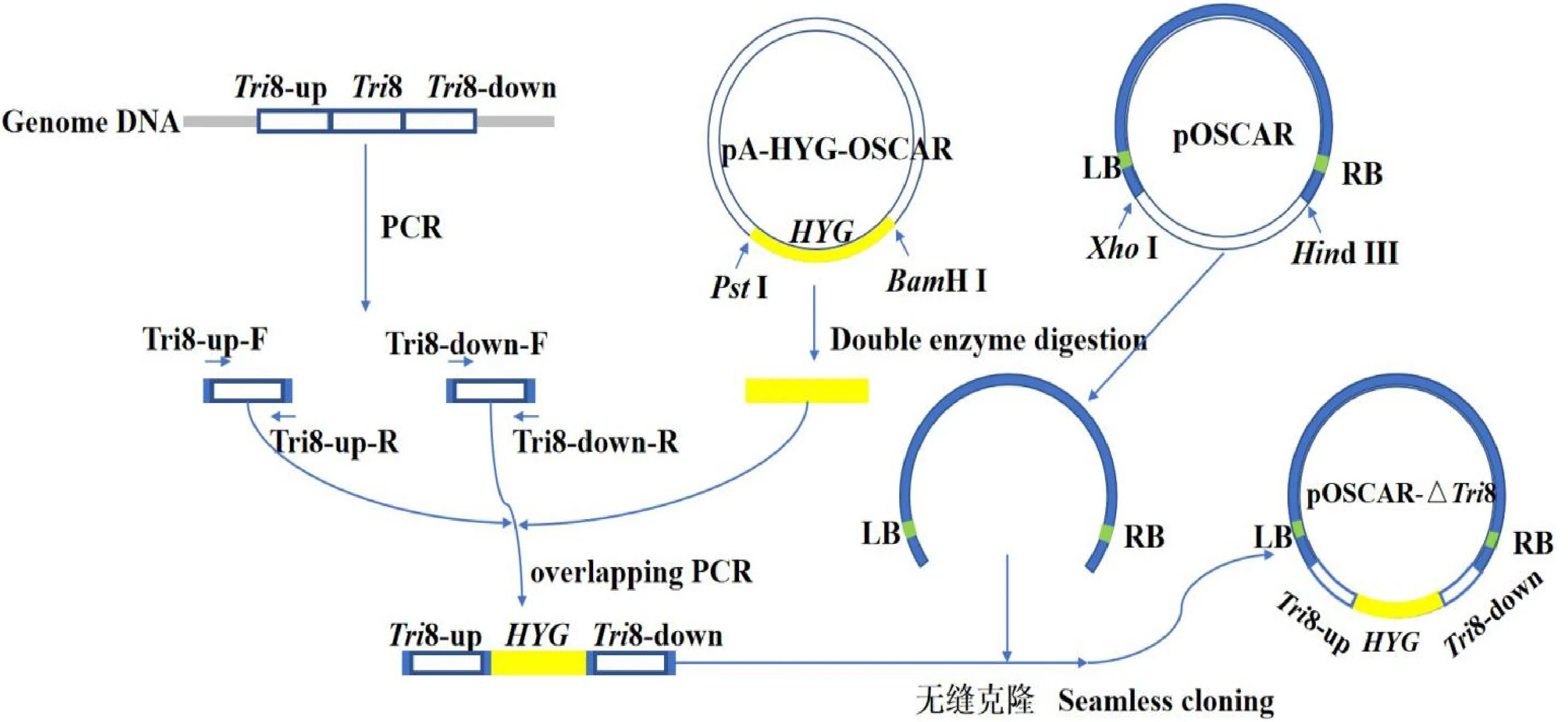
圖1 基因敲除載體pOSCAR-△ Tri8構建流程
2.2 基因敲除株系的篩選與鑒定結果
本研究利用農桿菌介導的遺傳轉化方法將敲除片段導入禾谷鐮刀菌PH-1中,利用同源重組機制將原有的Tri8置換為潮霉素抗性基因HYG。首先利用電激法將敲除載體pOSCAR-△Tri8導入農桿菌EHA105中,將其與鐮刀菌PH-1的孢子進行孵育,然后置于含有潮霉素的PDA平板進行篩選。提取陽性菌斑DNA,分別利用潮霉素抗性基因特異引物HYG-F/R和Tri8基因特異引物Tri8-F/R進行PCR擴增驗證,能擴增出HYG對應片段而不能擴增出Tri8對應片段的株系則為Tri8基因敲除株系PH-1-△Tri8。選取20個菌株提取基因組DNA進行PCR檢測,結果顯示大部分樣品均擴增出896 bp的潮霉素抗性基因特異條帶(圖2A)。但候選敲除菌株樣品6和18菌株菌絲較少,未成功提取其基因組DNA,故未成功擴增出896 bp的潮霉素抗性基因特異條帶。結果表明,除了野生菌株PH-1外,其他候選敲除菌株樣品中均未擴增出Tri8基因對應的1 388 bp特異條帶(圖2B),說明候選敲除菌株中(除樣品6和18)的Tri8基因已被HYG基因替換。

A:HYG基因擴增結果; B: Tri8基因擴增結果; M:1 kb DNA Ladder; 1:野生型菌株PH-1; 2~21:候選敲除菌株。
2.3 基因敲除株系的產毒類型檢測結果
按照材料與方法1.2.3所示流程對野生型菌株PH-1和Tri8基因敲除株系進行產毒培養,利用高分辨離子淌度液質聯用儀對粗提液的組分進行定性分析。發現野生型菌株中出峰時間為 4.97~5.00 min 的DON為最主要成分,同時存在少量的3-AcDON或15-AcDON,其出峰時間為5.98~6.00 min(圖3A);Tri8敲除株系中3-AcDON或15-AcDON含量最高,含有少量DON,在出峰時間6.48~6.51 min處出現第三個主峰,依據相對分子質量判斷此峰為3,15-diAcDON。與野生型菌株相比,Tri8基因敲除株系不同菌株的DON相關組分含量變化一致,均出現一定程度DON前體3,15-diAcDON的累積。
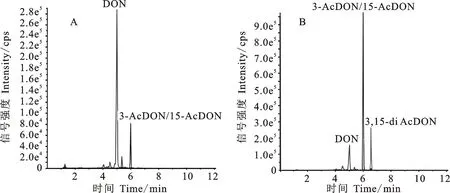
A:野生型菌株PH-1; B: Tri8基因敲除菌株。
3 討 論
由于赤霉菌毒素會對人畜健康造成嚴重威脅,因而赤霉菌毒素生物合成、分泌及調控機制一直是赤霉病研究的熱點[24]。Tri8編碼一種分泌型酯酶,可將禾谷鐮刀菌合成并轉運至胞外的毒素前體3,15-diAcDON上的乙酰基水解移除,形成只包含一個乙酰基的衍生物15-AcDON或3-AcDON[19],后者可自發或在其他病原菌或宿主酯酶作用下移除另一個乙酰基,最終形成DON。劉楊楊等[21]研究表明,PH-1菌株產毒類型主要為15-AcDON,即其Tri8編碼酯酶可催化雙乙酰前體3位乙酰基的水解。本實驗結果顯示,野生型菌株PH-1的毒素組分主要為DON,少量為 3/15-AcDON,而非理論上的以15-AcDON為主。表明在我們所給的實驗條件下[25],大部分15-AcDON可進一步自發或在其他酯酶催化下水解剩余的15位的乙酰基形成DON。實驗結果同時顯示,Tri8基因敲除株系產生的毒素以3/15-AcDON為主,并存在少量DON和3,15-diAcDON,而非理論上以3,15-diAcDON前體為主。考慮到野生型對照中大部分15-AcDON會因為自發或在其他病原菌或宿主酯酶作用下移除另一個乙酰基轉化為DON,因而突變體中大部分3,15-diAcDON被轉化為3/15-AcDON也在情理之中。無論如何上述結果都能說明,Tri8基因敲除會導致雙乙酰毒素前體的累積。
Tri8酯酶催化3,15-diAcDON乙酰基水解,是DON毒素合成和活化的重要步驟,要闡明該酶的催化機制首選需要制備出該酶足夠的底物3,15-diAcDON。本研究創制Tri8基因缺失突變體的初衷就是為了讓3,15-diAcDON在培養基中積累,然后抽提、分離制備獲得3,15-diAcDON。從已有的結果來看,Tri8基因的敲除的確可以導致3,15-diAcDON的累積,但大部分又自發轉化為3/15-AcDON。在后續研究中應進一步優化培養條件,減少3,15-diAcDON的自發轉化,實現3,15-diAcDON的大量累積,為后續Tri8基因結構和功能研究奠定基礎。
4 結 論
本研究利用重疊PCR和無縫克隆技術構建了禾谷鐮刀菌Tri8基因敲除載體pOSCAR-△Tri8,并利用農桿菌介導的轉化篩選鑒定出Tri8
