一種CRISPR-Cas9介導的擬南芥高效基因編輯系統的構建與應用
2020-08-04 09:09:22張成何明亮汪威徐芳森
中國農業科學 2020年12期
張成,何明亮,汪威,徐芳森
(華中農業大學作物遺傳改良國家重點實驗室,武漢 430070)
0 引言
【研究意義】通過基因編輯手段獲得基因敲除材料,對于研究基因在植物中的生物學功能具有重要的意義。而獲得高編輯效率的CRISPR-Cas9系統能加速獲得基因敲除材料的進程。【前人研究進展】在植物基因功能研究中,靶向基因編輯技術有著十分重要的作用,特別是在目標基因與其同源基因同源性較高的情況下很難通過其他途徑獲得想要的突變體。近些年,基于細菌免疫系統中CRISPR(clustered regularly interspaced short palindromic repeats)-Cas(CRISPR associated)的靶向基因編輯技術席卷了整個生物基因組研究領域[1-2]。常用的CRISPR-Cas9系統由兩部分組成:帶有核定位信號的Cas9和由21 bp靶序列(tracrRNA)的CRISPR(crRNA)構成的sgRNA(synthetic guide RNA)。其中,由sgRNA轉錄而成的非編碼RNA與帶有核定位信號的Cas9蛋白結合形成復合體,在PAM(protospacer-adjacent motif,Cas9系統中為NGG)位點上游3 bp位置切割DNA產生DNA雙鏈切割(double-stranded breaks,DSBs),繼而發生同源重組修復(homology-directed repair,HDR)或非同源重組修復(nonhomologous end joining,NHEJ),而容易出錯的NHRJ修復方式導致在切割位點附近發生核苷酸的隨機缺失,插入或者替換[1,3]。目前,CRISPR-Cas9系統已經成功在多種植物中得以應用,包括擬南芥、水稻、玉米、番茄、小麥、油菜和煙草等[4-11]。目前,在擬南芥中主要是利用農桿菌蘸花的方法,通過T-DNA插入到擬南芥卵細胞或者胚胎干細胞中的基因組獲得擬南芥轉基因植株。在之前的擬南芥CRISPR-Cas9研究中,主要是利用花椰菜花葉病毒(cauliflower mosaic virus,CaMV)35s啟動子啟動Cas9的表達,但發現擬南芥中的編輯效率較水稻下降很多,且發生基因編輯的T1代大部分為嵌合體。這主要由于35s啟動子在植物的胚胎形成過程中表達量不高,而是在營養器官中高表達量的原因[8,11]。因此,為了提高擬南芥中基因編輯的效率以及遺傳穩定性,選用合適的啟動子啟動Cas9的表達顯得尤為重要。目前已經有一些研究人員使用了在擬南芥卵細胞中表達水平高的啟動子來啟動Cas9的表達,比如EC1.1/EC1.2啟動子[12]、DD45和SPL啟動子[13]、YAO啟動子[14]、INCURVATA2啟動子[15]、APETALA1啟動子[16]以及RPS5A和WOX2啟動子[17]。【本研究切入點】擬南芥CRISPR載體的編輯效率大多較低,且從T1代編輯株系的鑒定到T3代純合突變且無Cas9材料的獲得過程耗時費力,造成嚴重的時間、人力和資源的浪費。創建擬南芥高效的CRISPR編輯系統有利于加速基因敲除材料的獲得,且節省時間和成本。【擬解決的關鍵問題】本研究通過構建一個高效CRISPR-Cas9系統用于擬南芥的基因編輯,并用擬南芥木葡聚糖內糖基轉移/水解酶基因TOUCH 4(TCH4)對該基因編輯系統進行驗證。采用DsRed2紅色熒光篩選標記篩選陽性植株及無Cas9植株。通過對測序峰圖的分析對編輯結果進行解碼,以便降低獲得穩定遺傳的無Cas9突變體材料的時間、人力和財務成本。
1 材料與方法
1.1 材料
擬南芥為Columbia-0(Col-0)生態型,生長條件為長日照(16 h光照/8 h黑暗),環境溫度為22℃。
1.2 擬南芥CRISPR-Cas9載體改造
擬南芥RPS5A編碼一個核糖體蛋白,從胚胎發育早期到后期所有的生長發育階段都表現為持續的高效表達[18-19]。使用的原始載體為pKSE401(由陳其軍教授課題組提供),為了構建pRSE-WH載體,首先使用引物pKSE-F:5′-GCGGGACTCTGGGGTTCG-3′和pKSE-R:5′-GCGGGACTCTGGGGTTCG-3′,用寶生物公司的primer STAR MAX高保真酶擴增不包含35S啟動子和潮霉素抗性的pKSE載體片段,同時以pBinGlyRed3的質粒為模板,用引物RED-F:5′-GCCAACATGGTGGAGGAGGAGTCCACCATGGT AGATCTG-3′和RED-R:5′-ACCCCAGAGTCCCGCT CAGGGTACCAGGAACAGGTG-3′擴增紅光蛋白片段,利用Infusion(TaKaRa)將2個片段連接,轉化后得到pRSE401載體。以pRSE401載體為模板,使用引物pRSE-F:5′-CTCGACCTCAACACAACATATA C-3′和pRSE-R:5′-GACCTGCAGGCATGCAAGC-3′,擴增得到不包含35S啟動子的pRSE401載體片段。利用擬南芥基因組DNA為模板,使用引物AtRPS5A-F:5′-GCATGCCTGCAGGTCCTCAACTTTTGATTCGCT ATTTGC-3′和AtRPS5A-R:5′-TGTGTTGAGGTCGAG GGCTGTGGTGAGAGAAACAG-3′擴增得到AtRPS5A啟動子片段,使用Infusion連接pRSE401載體片段和AtRPS5A啟動子片段,得到pRSE-WH載體。
1.3 TCH4的CRISPR載體構建
擬南芥TCH4(AT3G11940,XTH22)屬于木葡聚糖內糖基轉移/水解酶(xyloglucan endotransglucosylase/hydrolase,XTH)家族中的一員[20],具有2種催化活性:1)內切木葡聚糖;2)將內切產生的木葡聚糖還原端連接到另一個木葡聚糖的非還原端。XTH一方面可以剪切細胞壁中的木葡聚糖交聯網絡引起細胞壁的松弛,一方面可以參與細胞壁新組分的組裝,通過對細胞壁結構的調節參與對植物生長發育的調節[21-26]。利用CRISPR-P 2.0(http://crispr.hzau.edu.cn)對擬南芥TCH4設計2個靶點,靶點一序列為:5′-CCTTTCACTGCTTCTTACCG-3′,靶點二序列為:5′-GGATTGGAATCCAGAACCAG-3′。在引物的上游添加接頭:5′-ATTG-3′,在引物的下游添加接頭:5′-AAAC-3′。用于基因編輯第一個靶點的引物為:F:5′-ATTGCCTTTCACTGCTTCTTACCG-3′,R:5′-AAA CCGGTAAGAAGCAGTGAAAGG-3′。用于基因編輯第二個靶點的引物為:F:5′-ATTGGGATTGGAATCCA GAACCAG-3′;R:5′-AAACCTGGTTCTGGATTCCAA TCC-3′。采用25 μL退火體系(5 μL 10 mmol·L-1的上游引物,5 μL 10 mmol·L-1的下游引物,15 μL去離子水),退火程序為95℃ 3 min,95℃—16℃每20 s下降1℃,16℃保存,得到帶有黏性末端的靶序列二聚體。酶切連接體系為靶序列二聚體2 μL、pRSE-WH 2μL、10×NEB T4 buffer 1.5 μL、BsaⅠ(NEB)1 μL、10×BSA 1.5 μL、T4連接酶(NEB,高濃度)1 μL,去離子水補充至15 μL。反應程序為37℃ 5 h,50℃ 5 min,80℃ 10 min。將連接產物轉化大腸桿菌,送公司測序鑒定正確后,提取質粒轉化農桿菌。
1.4 擬南芥轉化以及陽性植株獲得和篩選
采用沾花法轉化擬南芥[27],收獲T1代種子之后,篩選標記為紅色熒光蛋白DsRed2,在綠色激發光下發射光為紅色,使用體式熒光顯微鏡篩選陽性植株。
1.5 編輯鑒定
采用CTAB法[28]提取T1代陽性植株的葉片基因組DNA,使用引物:TCR-F:5′-ATGGCGATCACTTAC TTGCTTC-3′和TCR-R:5′-TACTCTCTCTATGCAGCT AAGCAC-3′進行PCR擴增,測序檢測T1代植株編輯情況。
2 結果
2.1 擬南芥高效CRISPR-Cas9載體改造
首先,用DsRed2紅色熒光蛋白替換了原始的潮霉素抗性基因(圖1-A)。然后,利用擬南芥中RPS5A的啟動子替換pKSE401載體中的CaMV35s啟動子,從而達到在擬南芥胚胎發育早期進行基因編輯的目的。從擬南芥Col-0基因組DNA中擴增得到長度為1 659 bp的RPS5A的啟動子,將其整合到pRSE-WH載體中(圖1-A)。在pRSE-WH中,sgRNA由AtU6-26啟動子啟動,而在sgRNA和U6-26啟動子中間包含2個BsaⅠ酶切位點,可直接一步將20 bp的靶序列連入載體。替換后的紅色熒光蛋白可直接用于陽性植株的篩選以及后期無Cas9植株的篩選。
2.2 TCH4的CRISPR載體構建
TCH4的CDS全長為855 bp,基因結構包括2個外顯子和1個內含子。使用CRISPR-P 2.0(http://crispr.hzau.edu.cn)對擬南芥TCH4設計2個靶點。第一個靶點位于第二個外顯子上,第二個靶點位于第一個外顯子上(圖1-B)。在20 bp靶序列正向序列及反向互補序列的5′端添加接頭序列,通過變性-復性形成帶有黏性末端的二聚體,而pRSE-WH載體經過BsaⅠ酶切形成帶有粘性末端的線性化載體片段,利用T4連接酶連接即可將靶序列整合到pRSE-WH載體中。將帶有靶點的2個載體分別命名為TCR1和TCR2。
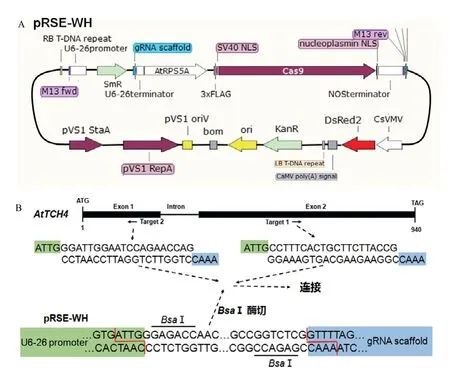
圖1 CRISPR/Cas9載體圖譜和TCH4的靶點示意圖Fig.1 CRISPR/Cas9 vectors and target mapping of TCH4 gene
2.3 紅光篩選
使用DsRed2作為篩選標記,用于陽性篩選。組成型表達的DsRed2蛋白,使得擬南芥種皮在綠光激發下發出紅光,區別于其他陰性種子(圖2)。將T1代紅色的陽性種子播種(圖2-A),收獲得到T2代種子,大部分的T2代種子中,紅光種子與非紅光種子的比例近似3﹕1(紅光種子與非紅光種子分別為65和28顆,圖2-B),符合分離定律。T2代挑選紅色種子繁種得到T3代不同植株種子中,三分之一的幾率得到純合紅色種子(圖2-C)。而T2代非紅光種子后代全部為非紅光種子(圖2-D)。
2.4 編輯檢測
從T1代植株中挑選40株TCR1和19株TCR2,提取植株葉片DNA,對TCH4基因組片段進行PCR擴增,測序檢測編輯結果。根據測序峰圖手動解碼編輯結果,結果發現,編輯類型可以分為4種:無編輯、純合編輯、雜合編輯以及雙等位編輯(圖3)。在40株TCR1植株中,發生編輯的有32株,編輯效率達到80%,19株TCR2植株中,全部發生編輯,100%編輯效率。通過對不同的編輯類型進行統計,發現59株T1代陽性植株中,無編輯8株(13.56%)、純合編輯9株(15.25%)、雙等位編輯40株(67.80%)和雜合編輯2株(3.39%)(圖4)。將每條染色體的編輯事件單獨分析,118次編輯事件中,18次沒有編輯,占比15.25%;45次單堿基插入,占比38.14%;1次多堿基插入,占比0.85%;11次單堿基缺失,占比9.32%;43次多堿基缺失,占比36.44%(表1)。其中多堿基缺失中,從幾bp到幾十bp均有發生,甚至幾百bp缺失。在單堿基插入事件中,TCR1中只存在2種事件,4次A堿基插入和28次T堿基插入,而TCR2中4種堿基插入均有發生,5次A堿基插入,2次T堿基插入,1次C堿基插入和5次G堿基插入。
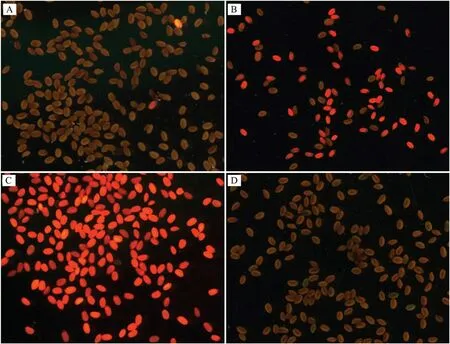
圖2 陽性植株的篩選Fig.2 Screening of positive plants
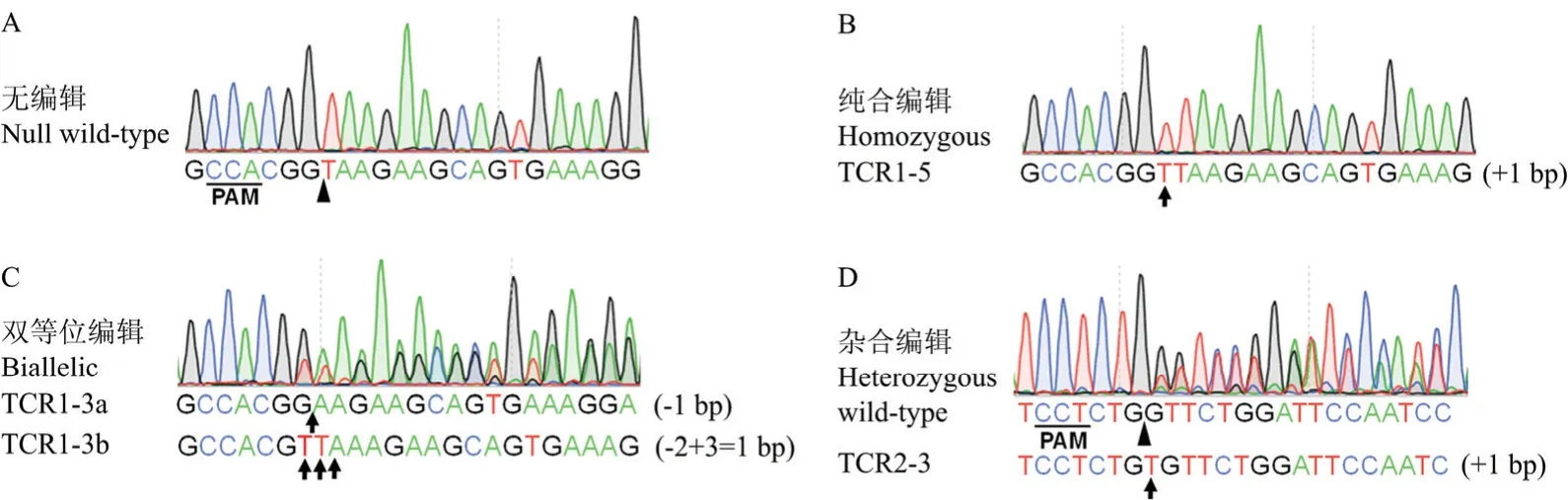
圖3 T1代植株的編輯類型Fig.3 Genotypes of the edited T1 plants
2.5 T2代編輯分析
在T1代發生純合編輯以及雙等位編輯的株系中選擇了無紅光種子進行繁種,并對T2代植株編輯情況進行測序檢測。結果顯示,TCR1-27株系T1代檢測結果為T堿基插入的純合編輯,所測序的6個T2代單株均為純合的T堿基插入。同樣,TCR1-10和TCR1-11的T1代均為雙等位編輯,所測序的6個T2代單株均顯示2條染色體中的突變都成功地遺傳到后代中(圖5)。
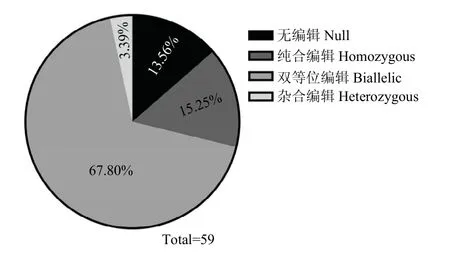
圖4 T1代植株各編輯類型所占比例Fig.4 Proportion of editing types in T1 plants
3 討論
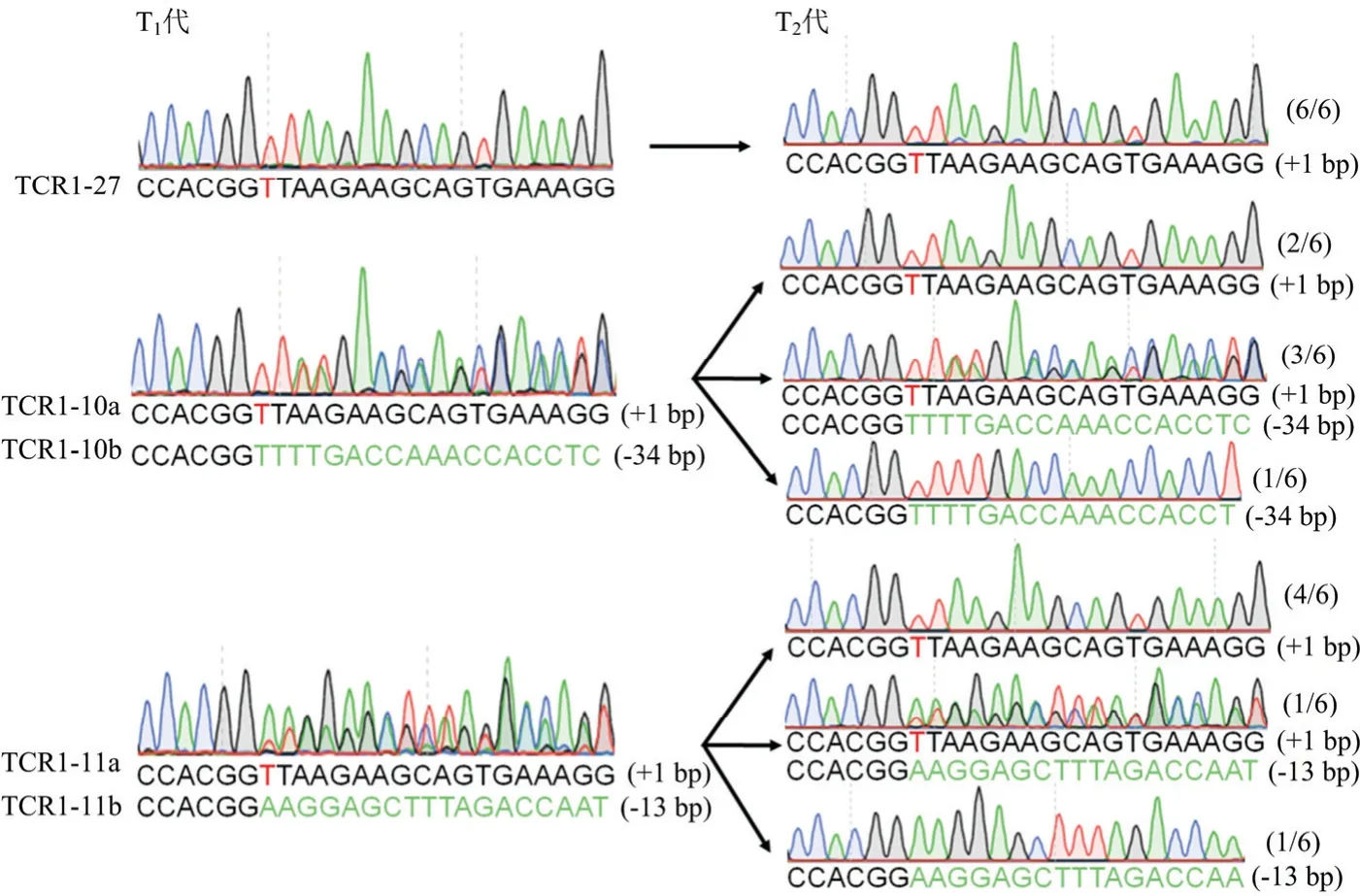
圖5 T1和T2代株系間的突變遺傳Fig.5 Mutation inheritance in T1 and T2 generation
以CRISPR為代表的基因編輯技術已廣泛應用于農作物基因功能驗證和農作物遺傳改良。目前,已研發多種CRISPR載體運用于單子葉和雙子葉植物的基因編輯[11-14,17,29]。植物營養遺傳實驗室嘗試了使用載體pKSE401在擬南芥和油菜中運用CRISPR技術對擬研究的候選基因進行基因編輯。但是發現在CRISPR實際操作中存在的兩個問題,一編輯效率低;二從T1代陽性植株篩選到T3代穩定遺傳株系的獲得,工作量大,成本高。而前者是主要的問題,因為一個高效的編輯系統能夠很大的降低后期的工作量和成本。植物營養遺傳實驗室前期的工作中使用載體pKSE401進行甘藍型油菜和擬南芥的編輯,其編輯效率較低,造成嚴重的時間、人力、資源的浪費。于是嘗試了對實驗室原有的CRISPR載體進行改造,試圖提高編輯效率。與一些植物采用外植體侵染-再生轉化植物不同的是,擬南芥遺傳轉化采用的是沾花法,農桿菌侵染的是雌性生殖器官中的胚囊。常用的35S啟動子在胚囊中或者在整個胚胎發育過程中表達量并不高,而這很有可能導致了擬南芥中編輯效率的下降。因此,本研究采用了一個在胚胎發育過程中持續高表達的RPS5A啟動子替換35S啟動子。
對TCH4的編輯結果顯示,RPS5A啟動子啟動Cas9的系統確實有著很高的效率,測序的59株轉基因陽性植株中編輯效率達到86%。而不同的靶點序列的編輯效率也有一定的差異,其中靶點一的編輯效率為80%,而靶點二的則是100%編輯。目前,CRISPR在水稻中的運用較好,編輯效率可以超過81%[11],在擬南芥中的編輯效率大多不超過40%[7,11,16]。而本研究改造的CRISPR編輯效率可高達86%。高的編輯效率可以很容易得到各種不同的突變類型,發生最多的是單堿基插入,占38.14%;其次是多堿基缺失,占36.44%;再次是單堿基缺失,占9.32%;而多堿基插入發生概率較低,118次編輯事件中只發生了一次。在這些不同的突變類型中,發現不同的靶點序列似乎對不同的突變類型有一定的偏好性。在TCR1的單堿基插入中,只發現了A/T插入,而且T插入占88%。但是TCR2的單堿基插入則4種堿基插入均有發現。導致這樣的結果可能與產生DNA雙鏈切割位點的堿基類型有關。TCR1的DNA雙鏈切割位點堿基為G-T,而TCR2的DNA雙鏈切割位點堿基為G-G。
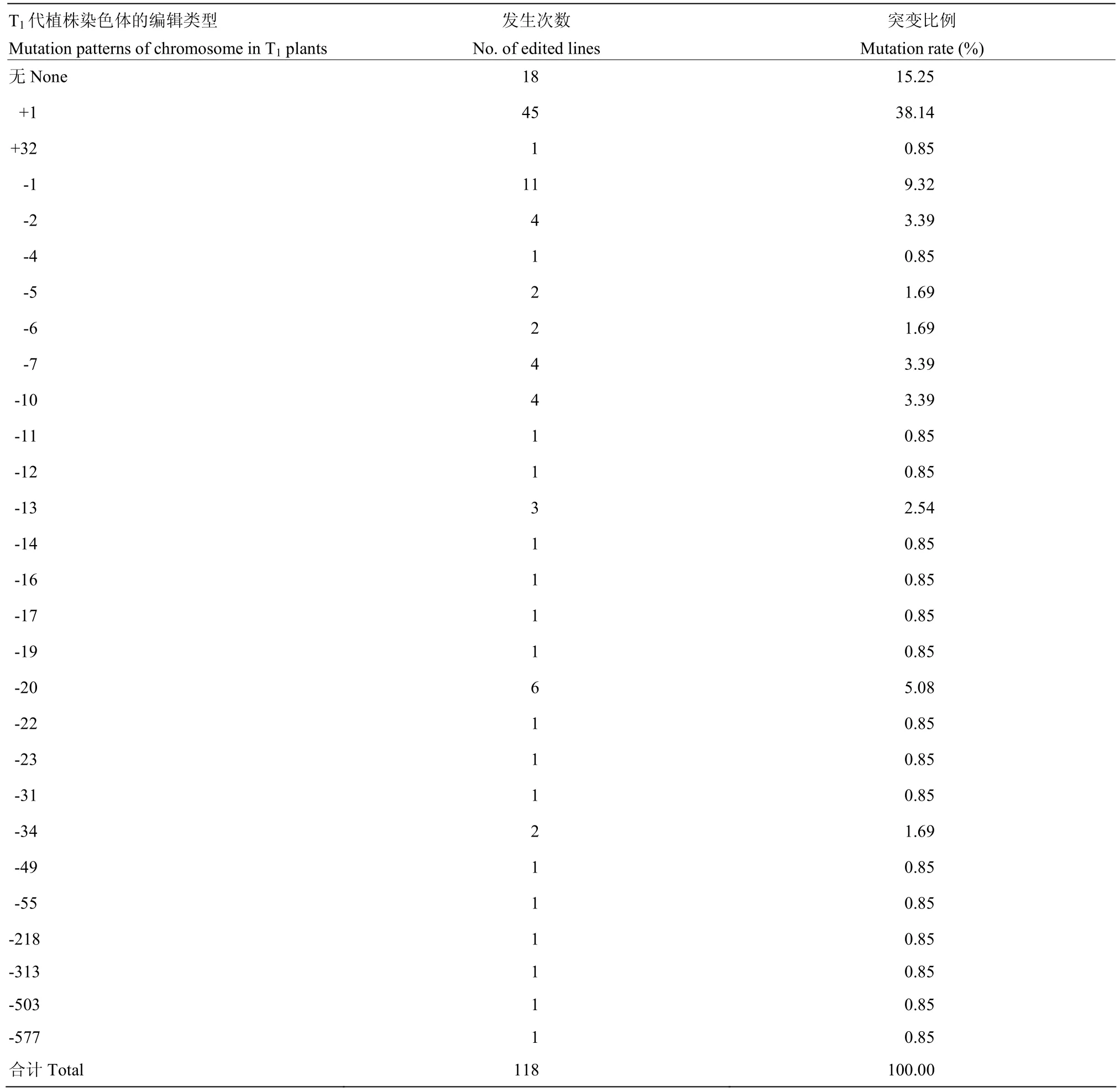
表1 TCH4 CRISPR T1代植株的編輯類型及比例Table 1 Mutation patterns and proportions TCH4 edited T1 plants
DsRed最早是從香菇珊瑚(Discosoma striata)中分離得到的一種紅色熒光蛋白[30]。DsRed的發射光譜峰值為583 nm,激發光譜峰值為558 nm。DsRed在應用過程中被發現存在一些問題,包括成熟緩慢,易形成四聚體,有一定毒性。第二代DsRed2的氨基末端進行了一些突變改造,使得其組織蛋白凝集和毒性下降,熒光基團成熟時間變短。DsRed2不僅可以像GFP一樣進行活體檢測和連續觀察,而且能消除植物本身的背景干擾,甚至在白光條件下也能檢測到紅光,因此,DsRed2可以作為一種可視標記應用于植物遺傳轉化[30-31]。在pRSE-WH中,采用DsRed2這種可視篩選標記。如果Cas9一直在植物中表達,它一方面可能會繼續對靶位點進行切割、編輯;另一方面脫靶的可能性也一直存在。因此,最終希望得到無Cas9且穩定遺傳的突變體。而Cas9基本上是與DsRed2連鎖的,所以通過篩選無紅光種子就能得到無Cas9的株系。在擬南芥CRISPR系統中,常用的抗生素篩選標記不適用于T2代無Cas9株系的篩選,而只能對T2代植株提取DNA,通過PCR擴增進行鑒定,理論上這會增加很大的工作量。而使用DsRed2可以很輕易的從T2代種子中篩選得到無Cas9的種子,并且能夠保證T3代種子中無Cas9的穩定遺傳。所以,在擬南芥CRISPR系統中DsRed2要明顯優于常用的抗生素篩選。此外,常用的解碼編輯結果的方法為對PCR產物進行克隆測序,并增加測序的單克隆數量獲得編輯的結果,這無疑是最可靠的方式之一,但這會增加大量的時間、人力和物力的消耗。本研究在了解解碼的機制后進行手動解碼:對目標基因的基因組片段進行PCR擴增,測序檢測編輯結果會出現2種峰型:單峰和套峰。其中,單峰表明擴增的PCR片段是單一的,可以很容易得到其中的DNA序列信息,而這種單峰的結果包括沒有發生編輯和純合編輯;套峰意味著PCR產物存在2個或者多個不同的DNA片段,會導致沒有那么容易獲得其中不同的DNA序列信息。可以通過以下幾點去解碼套峰中包含的信息:1)PCR過程中會導致不同DNA片段含量不一致,進而測序中表現出不同強度的信號值,可以通過這種信號強度的差異將不同的信息區分開;2)由于CRISPR-Cas9是基于DNA雙鏈切割進而發生DNA修復產生的編輯,所以它的特點是編輯會發生在切割位點附近或者向外擴散,而未編輯的位點則應該與參考序列一致,因此,即使在套峰中2個信號強度相近的DNA片段,依舊可以通過堿基間的序列信息推斷出其序列,解碼出其中一條信息之后,套峰的信息減去其中已知的信息,就能得到另一條的序列信息。基于以上兩點,對T1代陽性植株的測序信息進行解碼,選擇理想的編輯株系進行繁種,并在T2代中進行測序驗證,其結果等同于在T1代中進行克隆測序,且節省了大量的時間、精力和成本。
4 結論
構建了一個適用于擬南芥中基因編輯的高效CRISPR載體pRSE-WH,能夠簡便地獲得無Cas9且穩定遺傳的T3代突變體。
致謝:中國農業大學陳其軍教授提供了原始pKSE401載體;華中農業大學楊光圣教授課題組提供了體式熒光顯微鏡;華中農業大學汪社亮博士和英國The James Hutton Institute的Philip John White教授對文章的修改,在此表示感謝。
