醫藥中間體2-氯-3-氨基-4-甲基吡啶的合成研究
2020-08-23 07:37:26徐紅納才玉婷靳立國張朝立
當代化工 2020年7期
關鍵詞:工藝
徐紅納 才玉婷 靳立國 張朝立

摘????? 要: 以4-甲基吡啶為起始原料,經硝化、還原、氯化三步反應合成了醫藥中間體2-氯-3-氨基-4-甲基吡啶,總收率為68.3%,較原有的4-甲基吡啶縮減了合成步驟,收率提升了11%。
關? 鍵? 詞:2-氯-3-氨基-4-甲基吡啶;奈韋拉平;醫藥中間體;CAPIC
中圖分類號:TQ 253?????? 文獻標識碼: A?????? 文章編號: 1671-0460(2020)07-1277-04
Study on Synthesis of 2-Chlorine-3-amino-4-methyl Pyridine
XU Hong-na1, CAI Yu-ting1, JIN Li-guo2, ZHANG Zhao-li1
(1. Mudanjiang Medical College, Mudanjiang Heilongjiang 157011, China;
2. Heilongjiang No.1 Geological Exploration Institute, Mudanjiang Heilongjiang 157011, China)
Abstract: Using 4-methyl pyridine as the starting material, pharmaceutical intermediate 2-chloro-3-amino-4-methyl pyridine was synthesized via three steps of nitration, reduction and chlorination, the overall yield was 68.3%.Compared with the original process, the synthesis steps were decreased, and the yield was increased by 11%.
Key words: 2-chlorine-3-amino-4-methyl pyridine; Nevirapine; Pharmaceutical intermediate; CAPIC
奈韋拉平(Nevirapine),又名維樂命,分子式為C15H14N4O,化學名稱為11-環丙基-4-甲基-5,11-二氫-6H-二吡啶并[3,2-b:2,3-e][1,4]二氮雜卓-6-酮,是由德國Boehringer公司研制的一種HIV逆轉錄酶抑制劑,能夠有效地治療艾滋病[1-4]。2-氯-3-氨基-4-甲基吡啶(CAPIC)作為奈韋拉平重要中間體[5-8],其合成的2-氯-3-氨基-4-甲基吡啶中間體工藝與產品質量將直接影響奈韋拉平的成本與質量。
目前,2-氯-3-氨基-4-甲基吡啶CAPIC的合成路線主要包括2-氨基-4甲基吡啶法、氰乙酰胺路線法、丙二腈路線法、甲基吡啶路線法,2-氨基-4甲基吡啶法是Grozinger[9-10]報道的一種以2-氨基-4甲基吡啶法為原料先進行重氮化水解反應得到2-羥基-4-甲基吡啶,再經硝酸、硫酸硝化得到2-羥基-3-硝基-4-甲基吡啶,進一步利用五氯化磷和三氯氧磷氯化,最后加氫還原制備得到CAPIC(圖1)。該工藝轉化率較高,但硝化過程在鄰對位均容易發生,導致選擇性較差。
氰乙酰胺路線是以氰乙酰胺和乙酰乙酸乙酯為原料,在氫氧化鈉中發生環縮合,再與酰氯氯化、氰基水解、hofmann降解、加氫、氯化得到CAPIC[8,11](圖2),該路線原料易得,但反應步驟較長,收率較低。
丙二腈[12-14,7]路線法是以丙二腈和丙酮為起始原料經縮合、原酸三乙酯加成環合制備得到2-氨基-4-甲基-3-氰基吡啶,再經重氮化、氯化、氰基水解、hofmann降解得到CAPIC(圖3),該路線原料易得,但工藝較長收率不到10%。
甲基吡啶路線[15-17]是以甲基吡啶為起始原料經硝化得到N-硝基甲基吡啶,再經焦亞硫酸鈉作用得到3-硝基-4-甲基吡啶,進一步加氫將硝基還原為氨基,再經濃鹽酸-雙氧水氯化得到CAPIC(圖4),總收率為52%。該路線原料易得,收率相對較高,但合成步驟繁瑣,在工業化過程中設備投資與人工成本較高。
針對甲基吡啶路線現有工藝存在的問題,本文以甲基吡啶為原料,經硝化、還原、氯化三步直接合成CAPIC(圖5),能夠大幅度降低合成成本,減少設備投資。
1? 實驗部分
1.1? 試劑與儀器
1.1.1? 試劑
4-甲基吡啶,分析純,上海阿拉丁生化科技股份有限公司;硝酸,分析純,國藥集團化學試劑有限公司;濃硫酸,分析純,國藥集團化學試劑有限公司;氫氣,工業級,牡丹江市金瑞氣體有限公司;Pd/C,分析純,上海阿拉丁生化科技股份有限公司;氯氣,工業級,牡丹江市金瑞氣體有限公司。
1.1.2? 儀器
DLSB-40/80低溫冷卻循環泵,鞏義市予華儀器有限公司;DF-101S集熱式加熱磁力攪拌器,鞏義市予華儀器有限公司;PHSJ-6L型實驗室pH計,上海雷磁;GSHA-0.5高壓反應釜,威海鑫泰化工機械有限公司;DZF6500真空干燥箱,鞏義市予華儀器有限公司;GC-7890氣相色譜儀,美國安捷倫公司;5000 型高效液相色譜儀,美國 Varian 公司;Mercury plus 600 型核磁共振儀,美國 Varian 公,;WRS-1A 數字熔點儀(上海精密科學儀器有限公司) 。
1.2? 3-硝基-4-甲基吡啶的合成
向反應燒瓶中加入4-甲基吡啶,冷卻至0 ℃,緩慢地向反應器內滴加混合酸,控制反應溫度不超過10 ℃,滴加結束后在0 ℃下保溫3 h,再緩慢升至室溫保溫7 h,向反應體系中滴加氫氧化鈉將pH值調至7左右,用乙酸乙酯多次萃取水相,合并有機相,用無水硫酸鎂干燥旋蒸后得到黃色液體中間體1,純度95.6%[GC面積歸一化法:色譜柱 OV1701 ,升溫條件:起始溫度140 ℃,保持1 min,15 ℃/min升至250 ℃,保持8 min,氣化室溫度250 ℃,檢測器 FID,進樣量2μL,保留時間2.12 min]。1H-NMR(600 MHz,CD3OD),δ:9.51(s,1H, Py-2-H),7.49(s,1H, Py-5-H),8.92(s,1H, Py-6-H),2.32(s, 3H, CH3)。
1.3? 3-氨基-4-甲基吡啶的合成
向高壓釜內加入中間體,無水甲醇及5%Pd/C(g,),用氫氣置換高壓釜內的空氣,在70 ℃,向高壓釜內通入H2,反應至壓力開始升高時,繼續通入H2,保持壓力在0.4 MPa,保溫7 h,冷卻至室溫,過濾Pd/C,蒸出溶劑后,用堿液將pH值調至9左右,用乙酸乙酯萃取多次,有機相干燥后,蒸餾旋干得到淡黃色固體中間體2,純度為82.9%,1H-NMR(600 MHz,CD3OD),δ:7.89(s,1H, Py-2-H),7.66(s,1H, Py-6-H),6.98(s,1H, Py-5-H),4.85(s,2H, NH2), 2.16(s,3H, CH3)。
1.4? 2-氯3-氨基-4-甲基吡啶的合成
將中間體2及無水甲醇加入四口燒瓶中,在50 ℃條件下,向四口燒瓶內緩慢通入氯氣,控制反應溫度在50~60 ℃,用氣相色譜儀跟蹤反應進度,反應結束后,用30%氫氧化鈉溶液將體系pH值調至5左右,用二氯甲烷多次萃取,有機相合并后用無水硫酸鎂干燥,蒸出溶劑,重結晶提純后得到淡黃色固體3,純度99.7%,mp 69.3~70.4 ℃。1H-NMR(600 MHz,CD3OD),δ:7.51(d,1H, Py-6-H),6.97(d,1H, Py-5-H),4.81(s,2H,NH2),2.21(s, 3H, CH3)。
2? 結果與討論
2.1? 3-硝基-4-甲基吡啶合成工藝優化
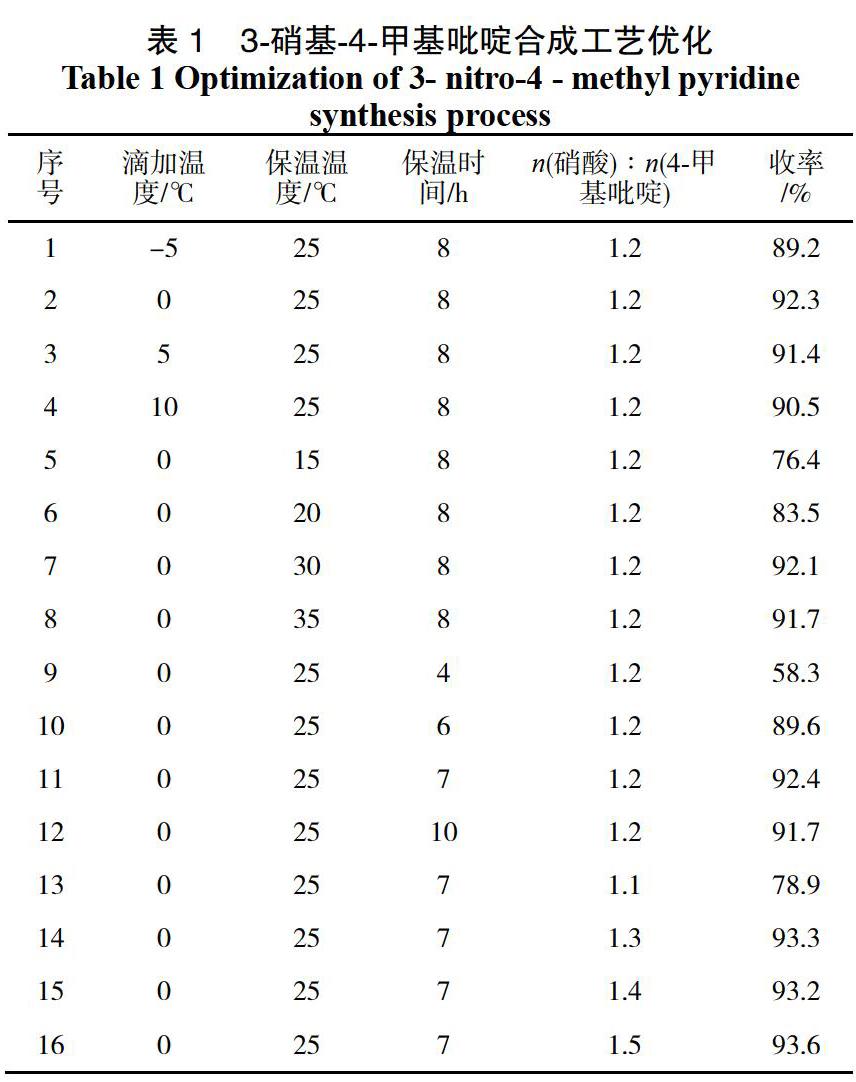
對3-硝基-4-甲基吡啶的合成工藝進行了單因素優化,分別探索了第一步合成過程中滴加溫度、保溫溫度、保溫時間、硝酸加入量對3-硝基-4-甲基吡啶收率的影響(見表1)。
表1為3-硝基-4-甲基吡啶合成工藝單因素優化實驗結果,表1中1-4為滴加溫度考察實驗,實驗結果可以看出在較低溫度下3-硝基-4-甲基吡啶收率較低,隨著溫度的升高收率有所增加,繼續提高溫度收率開始下降,這可能因為溫度過高導致副反應發生導致,最佳的滴加溫度為0 ℃。
表1中2、5-8項為保溫溫度對中間體1的收率影響。在相同保溫時間等條件下,保溫溫度較低時,由于反應能量不足收率較低,隨著溫度的升高中間體1的收率先增大后緩慢降低,當保溫溫度為25 ℃時,收率最大。表中2、9-12項為保溫時間對中間體1收率的影響,對結果進行分析,隨著反應時間的延長,反應收率逐漸提升,當反應時間為7 h時再增加反應時間收率變化不明顯,當反應時間大于10 h時收率略有下降,因此,最佳的反應時間為7 h,表1中11、13-16項為硝酸加入量對收率的影響,結果可以看出硝酸的加入也呈現出先增大后趨于平衡的趨勢,當n(硝酸)∶n(4-甲基吡啶)為1.3時收率最大為93.3%。
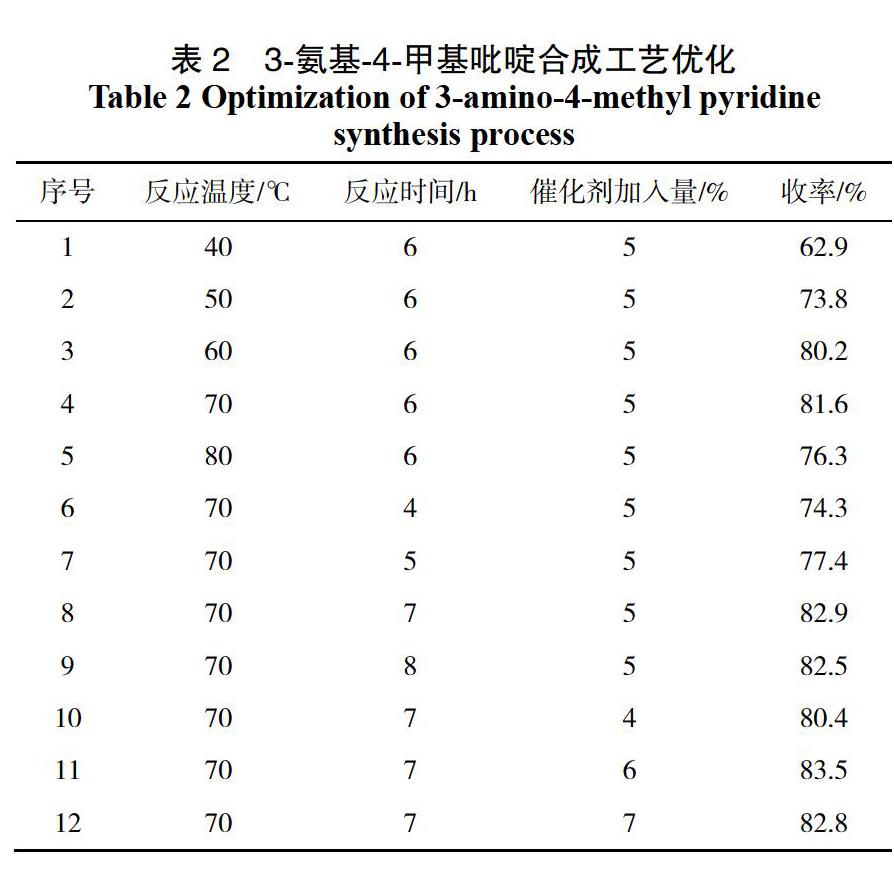
2.2? 3-氨基-4-甲基吡啶合成工藝優化
對3-氨基-4-甲基吡啶的合成工藝進行了單因素優化,分別探索了合成過程中反應溫度、反應時間、催化劑加入量對3-氨基-4-甲基吡啶收率的影響(見表2)。
表2為3-氨基-4-甲基吡啶合成工藝優化結果,其中1-5項為反應溫度單因素考察實驗,對結果分析可知,隨著反應溫度的增加3-氨基-4-甲基吡啶收率增大,當溫度高于70 ℃時,收率開始下降,因此最佳的反應溫度為70 ℃。表2中4、6—9項為反應時間優化實驗,從結果中可以看出隨著反應的進行,收率也逐漸增加,當反應時間7 h時收率趨于平穩,因此最佳的反應時間為7 h,表2中8、10-12項為Pd/C催化劑加入量單因素實驗,當催化劑加入量為6%時,中間體2收率最佳,該條件為最佳。
2.3? 2-氯3-氨基-4-甲基吡啶合成工藝優化
對2-氯3-氨基-4-甲基吡啶的合成工藝進行了單因素優化,分別探索了合成過程中反應溫度、鹽酸加入量、氯氣通入量對2-氯3-氨基-4-甲基吡啶收率的影響(見表3)。
表3為2-氯3-氨基-4-甲基吡啶合成工藝優化結果,表3中1-5項為反應溫度的優化結果,分別進行了30、40、50、60、70 ℃時CAPIC收率變化情況,隨著反應溫度的升高CAPIC的收率隨之增加,當溫度為50 ℃時,收率為67.5%,繼續提高溫度收率開始下降,最佳的反應溫度為50 ℃。表3中3、6-9項為體系濃度對收率影響,呈現出先增大后趨于平穩的趨勢,當濃度較大時容易產生副產品,因此,最佳的體系濃度為30%。表3中3、10—14項為氯氣加入量考察實驗,隨著氯氣加入量的增加收率增大,當加入量大于1.6∶1時,變化不明顯,因此最佳的加入量為n(氯氣)∶n(3-氨基-4-甲基吡啶)=1.6∶1。
3 ?結 論
1)以4-甲基吡啶為起始原料,經硝化、還原、氯化三步反應合成了醫藥中間體2-氯-3-氨基-4-甲基吡啶;
2)利用單因素法對2-氯-3-氨基-4-甲基吡啶合成工藝進行了優化,得到了最佳工藝條件,總收率為68.3%。
參考文獻:
[1] 謝威, 郭嘉. 奈韋拉平納米混懸液的制備及大鼠體內藥動學研究[J]. 中國藥師, 2015(1):23-27.
[2] 劉安, 葉江竹, 王鳳華,等. 188例HIV/AIDS病人接受包含奈韋拉平的ART后藥疹發生情況的臨床分析[J]. 中國艾滋病性病, 2015(4):273-274.
[3] 方世平, 榮玉萍, 高世成. 奈韋拉平+司他夫定+去羥肌酐相關性斯-約綜合征死亡1例[J]. 中國醫院藥學雜志, 2015, 35(12):1159-1160.
[4] 陳鐵龍, 駱名其, 馬智勇,等. 含奈韋拉平與依非韋侖抗反轉錄病毒治療后HIV-1耐藥突變比較[J]. 醫藥導報, 2016, 35 (7): 740-742.
[5] 甘立新, 周明華, 蹇鋒,等. 奈韋拉平中間體2-氯-3-氨基-4-甲基吡啶合成工藝研究進展[J]. 化學與生物工程, 2003, 20(s1):24-26.
[6] 許青青, 陳中元. 2-氯-3-氨基-4-甲基吡啶的合成[J]. 應用化工,2005, 34(2):72-75.
[7] 肖慶, 劉安昌, 譚珍友,等. 2-氯-3-氨基-4-甲基吡啶的合成[J]. 武漢工程大學學報, 2008, 30(4):33-35.
[8] 孟慶偉, 曾偉, 賴瓊,等. 奈韋拉平的合成[J]. 中國醫藥工業雜志, 2006, 37(1):5-7.
[9] GROZINGER K G, FUCHS V, HARGRAVE K D, et al. Synthesis of nevirapine and its major metabolite[J].Journal of Heterocyclic Chemistry, 2010, 32(1):259-263.
[10]GROZINGER K G, FUCHS V, HARGRAVE K D, et al. Synthesis of nevirapine and its major metabolite[J].Journal of heterocyclic chemistry, 1995, 32(1): 259-263.
[11]GROZINGER K G, HARGRAVE K D, Adams J. Method for preparing 3-amino-2-chloro-4-alkylpyridines:US, 5200522[P]. 1993-4-6.
[12]GROZINGER K G. Synthesis of 3-amino-2-chloro-4-methylpyridine from actone and ethylcyanoacetate:WO0043364[P]. 2000.
[13]徐志遠, 高桂祥, 邢友華. 氰乙酰胺和丙酮合成2-氯-3-氨基-4-甲基吡啶法, CN101565399[P]. 2009.
[14]GUPTON BF. Process for making 3-amino-2-chloro-4-methylpyridine: US, 2002052507[P]. 2002.
[15]劉剛, 孫林, 陳玉靜,等. 2-氯-3-氨基-4-甲基吡啶的合成[J]. 中國醫藥工業雜志, 2015, 46(6):571-573.
[16]劉田宇, 閻峰, 關瑾,等. 2-氨基-3-硝基-6-甲氧基吡啶的合成研究[J]. 當代化工, 2009, 38(5):450-452.
[17]梁海. 2-氨基-4,6-二甲氧基嘧啶的合成研究[J]. 當代化工, 2018, 47(9):59-61.
基金項目:黑龍江省自然科學基金(項目編號:H2016092);黑龍江省省屬高等學校基本科研業務費科研項目(項目編號:2017-KYYWFMY-0657)。
收稿日期:2019-08-01
作者簡介:徐紅納(1976-),女,河南省南陽市人,講師,碩士,2007年畢業于中國地質大學(北京)分析化學專業,研究方向:藥物化學技術。E-mail:13838782@163.com。
通訊作者:才玉婷(1981-),女,中級實驗師,研究方向:藥學。E-mail:joanna_688@126.com。
猜你喜歡
中國特種設備安全(2022年5期)2022-08-26 09:19:32
礦產綜合利用(2020年1期)2020-07-24 08:50:40
山東冶金(2019年6期)2020-01-06 07:45:54
收藏界(2019年2期)2019-10-12 08:26:06
世界農藥(2019年2期)2019-07-13 05:55:12
世界農藥(2019年2期)2019-07-13 05:55:10
模具制造(2019年3期)2019-06-06 02:11:00
山東工業技術(2016年15期)2016-12-01 05:30:59
銅業工程(2015年4期)2015-12-29 02:48:39
新疆鋼鐵(2015年3期)2015-11-08 01:59:52
