羅格列酮預處理對缺氧誘導腎小管上皮細胞壞死性凋亡的影響及其機制
2020-08-26 01:57:30席志楊覃遠漢潘競涂立蹇淑娟
山東醫藥 2020年23期
關鍵詞:模型
席志楊,覃遠漢,潘競,涂立,蹇淑娟
廣西醫科大學第一附屬醫院,南寧530021
腎間質纖維化(RIF)是各類腎臟病進展至終末期腎病的共同特征,其發生發展與腎小管上皮細胞(RTEC)損傷密切相關[1]。缺氧是最常見的RTEC損傷原因之一,可能導致RTEC的壞死性凋亡。壞死性凋亡作為一種具有壞死特征的細胞程序性死亡,是細胞死亡的重要形式[2,3]。抑制細胞壞死性凋亡可限制腎臟細胞的炎癥級聯反應,從而抑制腎臟細胞死亡,對腎臟具有一定的保護作用[4]。研究表明,過氧化物酶體增殖物激活受體γ(PPARγ)表達改變與細胞壞死性凋亡的發生密切相關[5]。2019年3月~2020年1月,本研究觀察了PPARγ激動劑羅格列酮預處理對缺氧誘導RTEC壞死性凋亡的影響,并探討其可能的機制。現報告如下。
1 材料與方法
1.1 材料 細胞:大鼠RTEC細胞株NRK-52E購于中國科學院上海生物細胞庫。主要試劑:高糖DMEM培養基、超濾胎牛血清均購于美國HyClone公司,羅格列酮試劑購于美國Sigma公司;兔單克隆一抗PPARγ購于美國SAB公司,兔單克隆一抗受體交互作用蛋白1(RIP1)、RIP3購于美國CST公司。
1.2 細胞分組處理 將NRK-52E細胞置于含10%胎牛血清及1%雙抗的高糖DEME培養基中,37 ℃、50 mL/L CO2培養箱中進行培養。待細胞融合80%時傳代,接種到新的培養瓶中;待細胞貼壁生長融合60%~70%時,更換新的培養基。將細胞隨機分為羅格列酮組、缺氧模型組及正常對照組。羅格列酮組給予15 μmol/L羅格列酮進行預處理,缺氧模型組及羅格列酮組置于含95% N2和5% CO2混合氣體的缺氧小室中,正常對照組置于37 ℃、50 mL/L CO2培養箱中,各組均培養48 h。
1.3 細胞超微結構觀察 將各組細胞消化離心后,棄去培養基,沿離心管管壁緩慢加入預冷的2.5%戊二醛固定液固定4 h。4 ℃條件下0.1 mol/L二甲砷酸鈉緩沖液清洗細胞3次,每次2 h;1%鋨酸溶液浸泡2 h,4 ℃條件下0.1 mol/L二甲砷酸鈉緩沖液清洗細胞2次,每次15 min。4 ℃條件下依次用50%、70%、80%乙醇脫水10 min,90%乙醇脫水15 min,純叔丁醇溶液脫水15 min。室溫條件下進行滲透、包埋過夜及固化。光鏡下用半薄切片機定位固化好的樣本,再用超薄切片機將樣本切成50 nm厚度的超薄切片,進行枸櫞酸鉛溶液染色。透射電鏡下觀察各組細胞超微結構。
1.4 細胞PPARγ、RIP1、RIP3蛋白表達檢測 采用Western blotting法。取各組細胞,提取總蛋白,BCA蛋白定量試劑盒測定蛋白濃度。4 ℃條件下,1 2000 r/min離心15 min,吸取上清;按照4∶1的比例加入蛋白上樣緩沖液,水浴煮沸變性,行十二烷基硫酸鈉-聚丙烯酰胺凝膠泳(SDS-PAGE),濕轉法轉膜至聚偏二氟乙烯膜(PVDF)。5%脫脂牛奶室溫下封閉1.5 h,加入PPARγ、RIP1、RIP3、β-actin特異性一抗(稀釋比例均為1∶1 000),4 ℃條件下孵育過夜,TBST溶液洗膜3次×10 min。加入二抗(稀釋比例均為1∶10 000),室溫條件下孵育1.5 h,TBST溶液洗膜3次×10 min。以發光液覆蓋PVDF膜,孵育1 min后,采用Carestrecm Image Station化學發光儀掃描PVDF顯影。采用Alpha View軟件檢測條帶灰度值,計算PPARγ、RIP1、RIP3蛋白相對表達量。
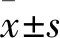
2 結果
2.1 各組細胞超微結構觀察結果 正常對照組細胞形態完整,線粒體無腫脹,細胞核無破裂,核仁無固縮。缺氧模型組及羅格列酮組均出現細胞腫脹,線粒體明顯腫脹,細胞核及染色質固縮;羅格列酮組線粒體腫脹、細胞核及染色質固縮現象均較缺氧模型組減輕。
2.2 各組細胞PPARγ、RIP1、RIP3蛋白相對表達量比較 缺氧模型組、羅格列酮組和正常對照組細胞PPARγ蛋白相對表達量依次升高、RIP1及RIP3蛋白相對表達量均依次降低,組間兩兩比較P均<0.05。見表1。
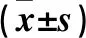
表1 各組細胞PPARγ、RIP1、RIP3蛋白相對表達量比較
2.3 PPARγ與RIP1、RIP3蛋白相對表達量的相關性分析結果 相關性分析結果顯示,PPARγ與RIP1、RIP3蛋白相對表達量均呈負相關關系(r分別為-0.608、-0.317,P均<0.05)。
3 討論
RIF是各類腎臟疾病進展至終末期腎病的共同特征,目前認為RIF的發生發展與RTEC損傷密切相關。凋亡和壞死是細胞死亡的兩種經典模式,而壞死性凋亡是一種不同于凋亡和壞死的新型細胞死亡途徑,是具有壞死特征的細胞程序性死亡。細胞凋亡的關鍵調控因子是Caspase[6],但有研究顯示,負責細胞凋亡的主要信號傳導通路是腫瘤壞死因子α(TNF-α) /腫瘤壞死因子受體(TNFR)受體信號通路;該通路被激活時,即便抑制Caspase也不能阻止細胞凋亡,而是使細胞轉移到一種具有壞死形態學特征的死亡模式[3,7]。Krysko等[8]將這種特殊的細胞死亡形式首次命名為壞死性凋亡。壞死性凋亡同時具有壞死的細胞質和凋亡的細胞核,主要變現為細胞腫脹,各細胞器如線粒體腫脹甚至空泡化,核染色質固縮邊聚,多呈月牙狀[9]。壞死性凋亡作為一種重要的細胞死亡形式,其調節、誘導及阻斷機制是一個復雜的過程,涉及一系列分子的表達及調控[3,10]。研究證實,壞死性凋亡主要由RIP1、RIP3活化介導,二者是壞死性凋亡發生的關鍵,也是其特異性標志[10,11]。抑制腎臟細胞的壞死性凋亡可以減輕炎癥因子表達、限制炎癥級聯反應放大,從而減輕腎臟細胞損傷,對腎臟具有一定的保護作用[4]。本研究結果顯示,缺氧模型組、羅格列酮組透射電鏡下可見線粒體明顯腫脹、細胞核及染色質固縮,兩組壞死性凋亡特異性蛋白RIP1、RIP3蛋白表達量均高于正常對照組,且缺氧模型組上述變化更顯著;提示缺氧可導致RTEC壞死性凋亡,羅格列酮預處理可減輕缺氧造成的RTEC壞死性凋亡。
PPARγ表達改變與壞死性凋亡的發生關系密切。在膿毒癥導致心功能不全大鼠模型中,激活PPARγ可減少心肌細胞的壞死性凋亡,從而提高膿毒癥大鼠的存活率;提示上調PPARγ可抑制心肌細胞的壞死性凋亡,減輕感染導致的心功能不全[5]。PPARγ表達異常與腎臟疾病的發生發展密切相關。研究發現,對缺血再灌注損傷大鼠應用羅格列酮可上調其PPARγ表達,并增強機體抗氧化能力,從而減輕腎臟組織損傷[12,13]。研究發現,活化氨基末端激酶通路可上調糖尿病小鼠胰島細胞及腎小球系膜細胞PPARγ表達,從而抑制細胞凋亡,在改善胰島素抵抗的同時減輕了腎臟損傷,提示PPARγ可能具有減輕腎臟損傷的作用[14,15]。本研究結果顯示,缺氧模型組和羅格列酮組PPARγ蛋白表達均顯著低于正常對照組,且缺氧模型組降低更明顯;提示羅格列酮減輕RTEC壞死性凋亡的機制可能與其上調PPARγ蛋白表達有關。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
網絡安全與數據管理(2022年1期)2022-08-29 03:15:20
導航定位學報(2022年4期)2022-08-15 08:27:00
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
新世紀智能(數學備考)(2021年9期)2021-11-24 01:14:36
成都醫學院學報(2021年2期)2021-07-19 08:35:14
新世紀智能(數學備考)(2020年9期)2021-01-04 00:25:14
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
光學精密工程(2016年6期)2016-11-07 09:07:19
