熱處理輝鉬礦光催化原位還原銀離子的研究
2020-10-17 08:14:24賈菲菲楊丙橋宋少先
硅酸鹽通報 2020年9期
袁 媛,賈菲菲,楊丙橋,邢 浩,宋少先
(1.武漢理工大學資源與環境工程學院,武漢 430070;2.武漢工程大學興發礦業學院,武漢 430073)
0 引 言
改革開放以來,我國電鍍等行業發展迅猛[1],但由于環境保護意識淡薄,導致產生的含銀廢水隨意排入河流。微量銀離子(Ag+)即可對水體生物特別是魚類造成巨大的危害,且對水環境造成嚴重污染,最終危害人類健康[2]。在“綠水青山就是金山銀山”的新時代,從含銀廢水中回收銀,不僅響應了新時代綠色發展的理念,同時具有十分重要的經濟價值和環保價值,是環境保護所面臨的重要課題之一[3-5]。目前常用的銀回收方法有金屬置換法、離子交換法、化學沉淀法和電解法[6],其中吸附法因成本低廉、操作簡單而引起廣泛關注[7]。活性炭[8]、活性炭纖維[9]和聚氨酯海綿[10]等材料因制備復雜、與Ag+絡合作用不大等缺點,難以應用于實際廢水處理中。根據皮爾遜軟硬酸堿理論[11]可知,Ag原子與S原子具有較強親和力,因此尋找富硫材料對Ag+的高效吸附回收十分重要。
二硫化鉬(MoS2)是由兩層六角排列的S原子夾雜一層同樣六角排列的Mo原子,S和Mo原子以共價鍵形式結合成經典的三明治結構,其表面具有豐富的S原子[12],可作為Ag+吸附回收的潛在材料。MoS2包括1T、2H和3T相三種晶體結構,塊體形態的MoS2也被稱作輝鉬礦,常見的天然輝鉬礦為2H半導體相,也是最為穩定的一種賦存狀態,在自然界中含量豐富[13]。同時輝鉬礦也是一種廣泛使用的光催化材料,與零帶隙的石墨烯不同,輝鉬礦隨片層厚度不同而使得帶隙可在1.29~1.90 eV之間調節,在可見光較寬范圍內均具有光電流響應[14]。當可見光照射到輝鉬礦表面時,光子可將位于MoS2價帶位的電子激發并使其躍遷到導帶位置,同時在價帶位置留下具有氧化性的空穴,這些光生電子可在輝鉬礦表面發生還原反應[14]。
考慮到Ag+/Ag0的還原電位約為0.789 eV[15],而輝鉬礦的導帶約為0 eV[14],可將Ag+直接還原成Ag單質便于回收。基于輝鉬礦的半導體性質光催化原位還原Ag+,不僅縮減了傳統Ag單質回收技術中的吸附-脫附-還原三個步驟,而且輝鉬礦可將Ag+直接還原成Ag納米顆粒。此外,相對于其他制備Ag單質顆粒的方法如電化學法、化學還原法和生還原法等,光化學還原法具有成本低、操作簡單且綠色環保的優點[15]。與目前已應用于光化學還原沉積貴金屬的材料例如二氧化鈦[16]、高分子殼聚糖[17]和聚乙烯吡咯烷酮[18]等材料相比,輝鉬礦因其飽含S原子、具窄帶隙半導體特性、價格低廉、來源廣泛等優點而引起廣泛關注[19]。然而天然輝鉬礦廣泛暴露的基面化學活性不大,導致活性點位較少,還原性能較差[20],因此如何進一步豐富輝鉬礦的活性位點是提升Ag+吸附回收效果的關鍵問題。
熱處理是一種增加活性位點及調節光吸收能力的最簡單方法之一,輝鉬礦經熱處理后將產生缺陷[21],從而大量暴露對Ag具有很強的絡合作用的不飽和S原子,因此通過熱處理輝鉬礦可用于提升Ag+的還原效果。本工作還考察了不同pH值、不同還原時間及不同Ag+初始濃度對還原效果的影響,并對其Ag+提升原因進行了分析,從而為高效回收Ag+提供一種新的思路。
1 實 驗
1.1 原材料
試驗中所用水均來自Millipore-Q設備(電阻率為18.2 MΩ·cm)過濾后的去離子水,且所涉及到的試劑溶液均為現配現用。試驗中使用的輝鉬礦均來源于廣西省梧州市南方礦業有限公司,在原子力顯微鏡(AFM)測試中所用的輝鉬礦為塊狀樣,首先用剪刀裁剪成0.05 cm×1 cm×1 cm的塊狀樣,再由3M膠帶撕掉表層,從而暴露出干凈的新表面以備測試。此外所有試驗均使用粒度大小均為+45 μm的輝鉬礦顆粒。實驗中所用到的試劑HNO3與AgNO3均來自國藥集團化學試劑有限公司,分析純。
1.2 制備熱處理二硫化鉬
稱取10.0 g輝鉬礦顆粒及一片塊狀輝鉬礦置于瓷舟中,放進馬弗爐中,在正常空氣條件下,保持10 ℃/min的升溫速率,由室溫升至400 ℃以后保持恒溫3 h,后自然降溫到室溫,用去離子水沖洗三次后過濾烘干備用。
1.3 樣品表征
試驗中用到的顆粒狀輝鉬礦未進行其他處理,直接用于測試。還原Ag+試驗完成后,經抽濾凍干后的樣品進行掃描電子顯微鏡及其能譜圖(SEM-Mapping)、X射線光電子能譜(XPS)、X射線衍射(XRD)、拉曼(Raman)等測試。利用D8 Advance X射線衍射儀對樣品進行物相分析,測試條件為Cu-Kα輻射源,掃描速度10°/min。利用ESCALB 250Xi光電子能譜儀對樣品進行化學元素狀態分析。利用Zeiss Gemini 300 SEM對樣品的形貌進行表征。使用INVIA Raman光譜儀對樣品結構進行表征。利用德國Bruker公司的MultiMode 8型原子力顯微鏡測試熱處理前后輝鉬礦微觀形貌,測試使用的是峰值力模式。利用Netzsch Sta 449F3同步熱分析儀對輝鉬礦的穩定性進行表征,測試條件為從常溫至1 000 ℃加熱,升溫速率10 ℃/min。利用美國 PerkinElmer公司的Lambda 750S型紫外可見近紅外分光光度計測試樣品的紫外可見光譜,研究熱處理前后輝鉬礦的光催化性能。利用日本島津(SHIMADUN)公司的AA-6880型原子吸收光譜儀測定溶液中Ag的含量。
1.4 原位還原銀離子
還原動力學試驗方法:稱取0.5 g天然輝鉬礦及熱處理輝鉬礦,放進1 L初始質量濃度為50 mg/L的硝酸銀溶液中,室內光條件下反應,在特定時間(0~600 min)通過0.22 μm濾膜過濾取2 mL溶液,將前1 mL丟棄,另1 mL稀釋,利用原子吸收光譜儀測量溶液中Ag+濃度。
初始濃度對焙燒輝鉬礦還原Ag+影響實驗:稱取0.5 g熱處理輝鉬礦,放進不同質量濃度的硝酸銀溶液中,體積為1 L,放置于恒溫振蕩器中,于室內光(即光照波長為可見光波段390~780 nm范圍)條件下反應,轉速為200 r/min,溫度25 ℃,24 h后取樣。
pH值對熱處理輝鉬礦還原Ag+影響實驗:稱取0.5 g熱處理輝鉬礦,放進初始質量濃度為50 mg/L,pH值分別為1.5、2、2.5、3、4和4.6的硝酸銀溶液中,放置于恒溫振蕩器中,室內光條件下反應,轉速為200 r/min,溫度25 ℃,24 h后取樣。
2 結果與討論
2.1 熱處理輝鉬礦結構表征
圖1所示為400 ℃熱處理前后輝鉬礦的XRD譜。由圖可知,輝鉬礦的XRD譜中出現了14.378°、29.027°、32.677°、39.539°、44.152°、49.788°、60.146°、70.144°,以及78.119°的衍射峰,天然2H型MoS2的JCPDS卡片序號為37-1492[22],上述位置的衍射峰分別對應MoS2的(002)、(004)、(100)、(103)、(006)、(105)、(008)、(108),以及(109)晶面。熱處理后輝鉬礦的XRD譜與天然輝鉬礦對比分析,發現熱處理后衍射峰幾乎一致,證明輝鉬礦的晶體結構未發生明顯變化。
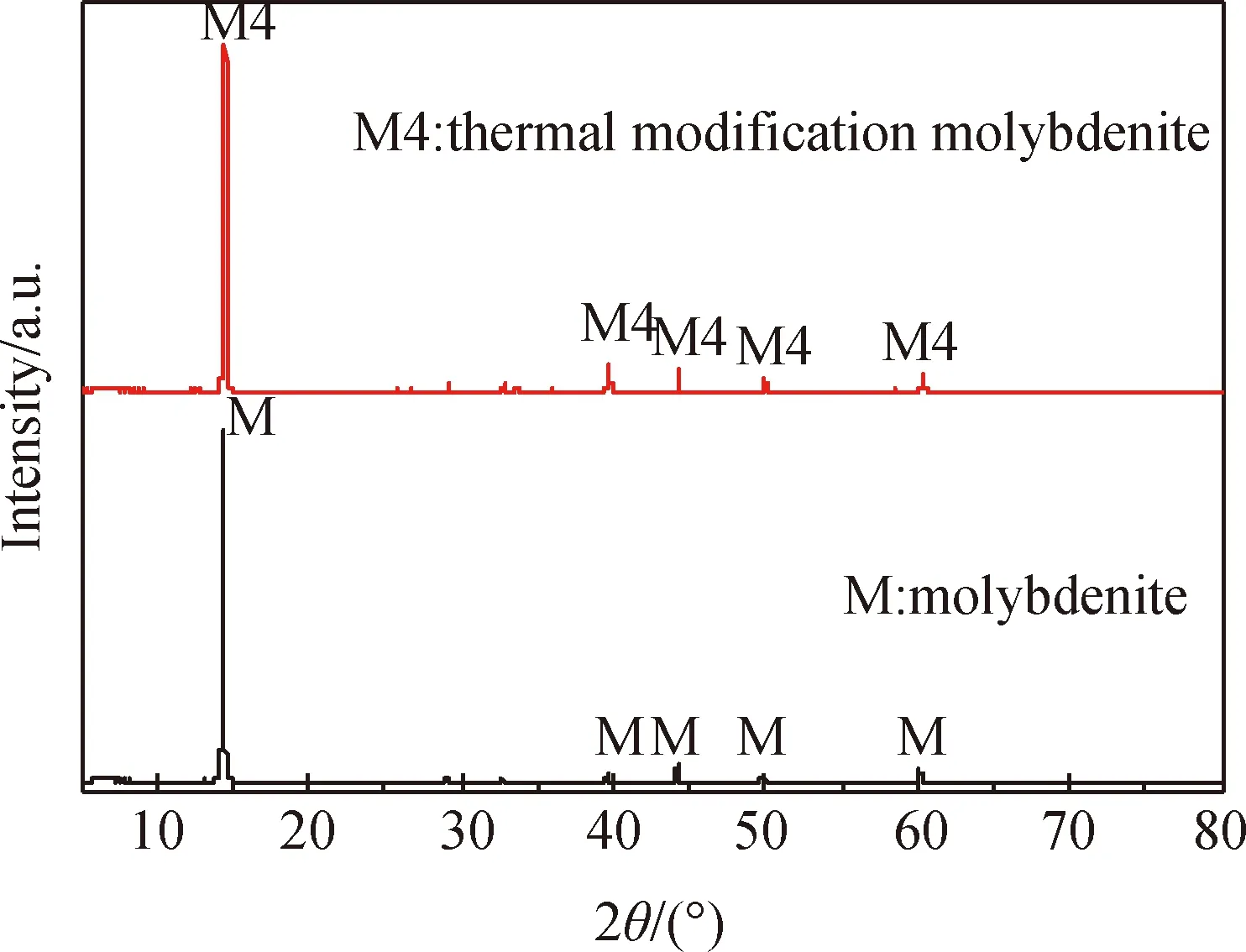
圖1 400 ℃處理后的輝鉬礦XRD譜Fig.1 XRD patterns of molybdenite after thermal modification at 400 ℃

圖2 輝鉬礦的熱重曲線Fig.2 TG curve of molybdenite
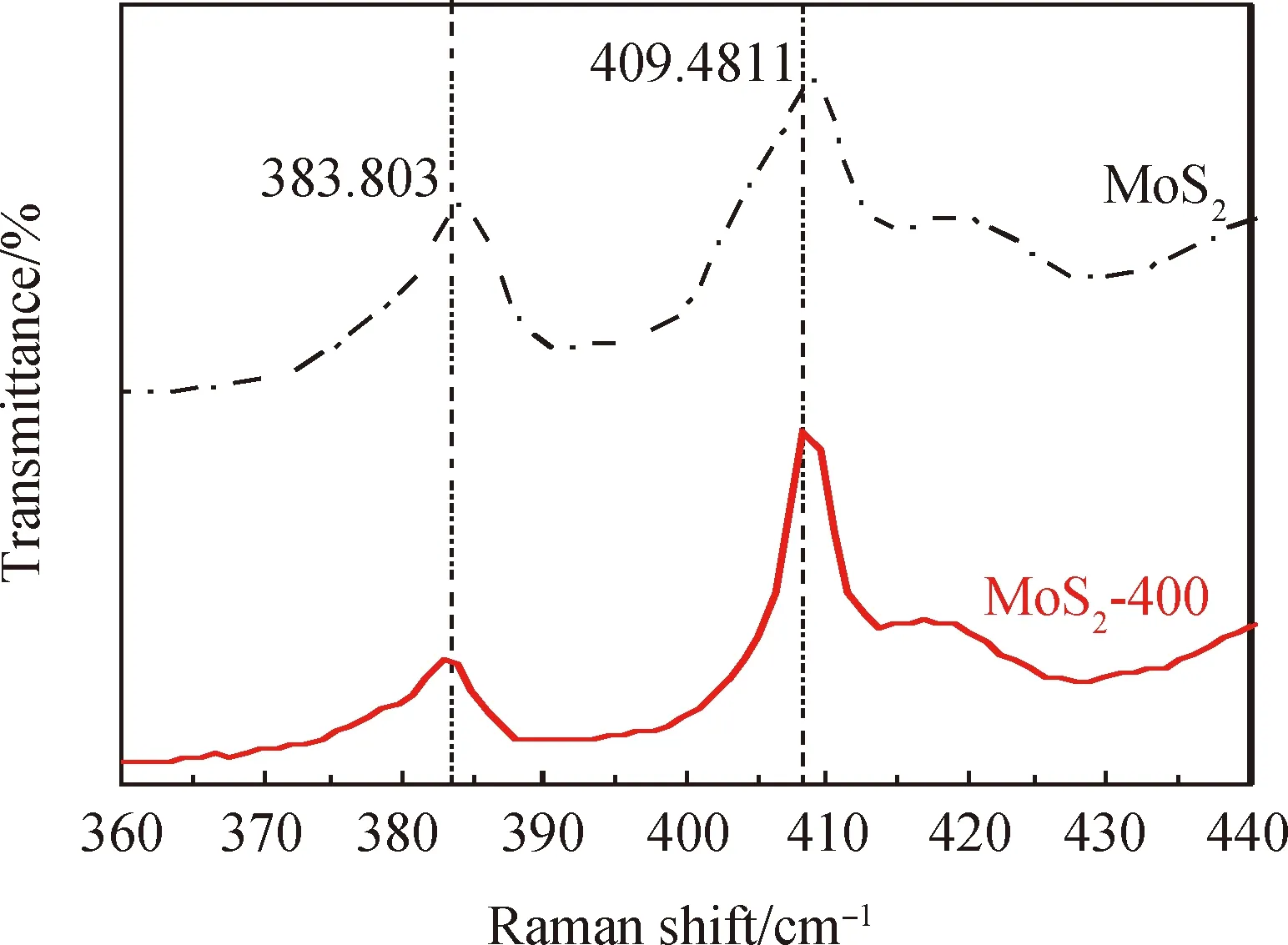
圖3 輝鉬礦熱處理前后的拉曼光譜Fig.3 Raman patterns of molybdenite before and after thermal modification
輝鉬礦的熱重曲線如圖2所示。由圖可知,在空氣氣氛下,400 ℃前,輝鉬礦由于少量水分的揮發導致0.43%的質量損失[23]。500 ℃至800 ℃之間,由于結構水的揮發,輝鉬礦質量損失了9.89%[24]。當溫度超過800 ℃后,輝鉬礦質量急劇下降,這是由于在高溫煅燒下,輝鉬礦氧化產生可揮發含硫氣體,在此過程中,輝鉬礦結構被嚴重破壞。當溫度達到1 000 ℃時,重量基本不再損失,剩余物質主要為灰分,含量大約為天然輝鉬礦的10%[25]。因此,在400 ℃的溫度下焙燒,輝鉬礦的整體結構不會被破壞,與前面XRD的分析一致。
輝鉬礦熱處理前后的拉曼光譜如3所示。從圖可以看出,輝鉬礦的兩個峰值分別為383.803 cm-1和409.481 cm-1,這兩個峰屬于輝鉬礦的特征拉曼峰[26]。經400 ℃熱處理后輝鉬礦特征峰的位置幾乎沒有變化,且兩個特征峰的間距基本不變,再次證明熱處理對輝鉬礦的晶體結構沒有太大的影響。
熱處理前后輝鉬礦表面的AFM照片如圖4所示。從圖中可看出輝鉬礦原礦表面十分光滑干凈,而經熱處理后輝鉬礦表面產生了部分三角形形狀的凹坑缺陷。據報道,缺陷邊緣多為不飽和S原子,在吸附催化等領域中均為反應活性位點[21],此結果說明熱處理過程可使輝鉬礦表面產生較多活性位點,從而有利于更多Ag+在熱處理輝鉬礦表面的吸附。
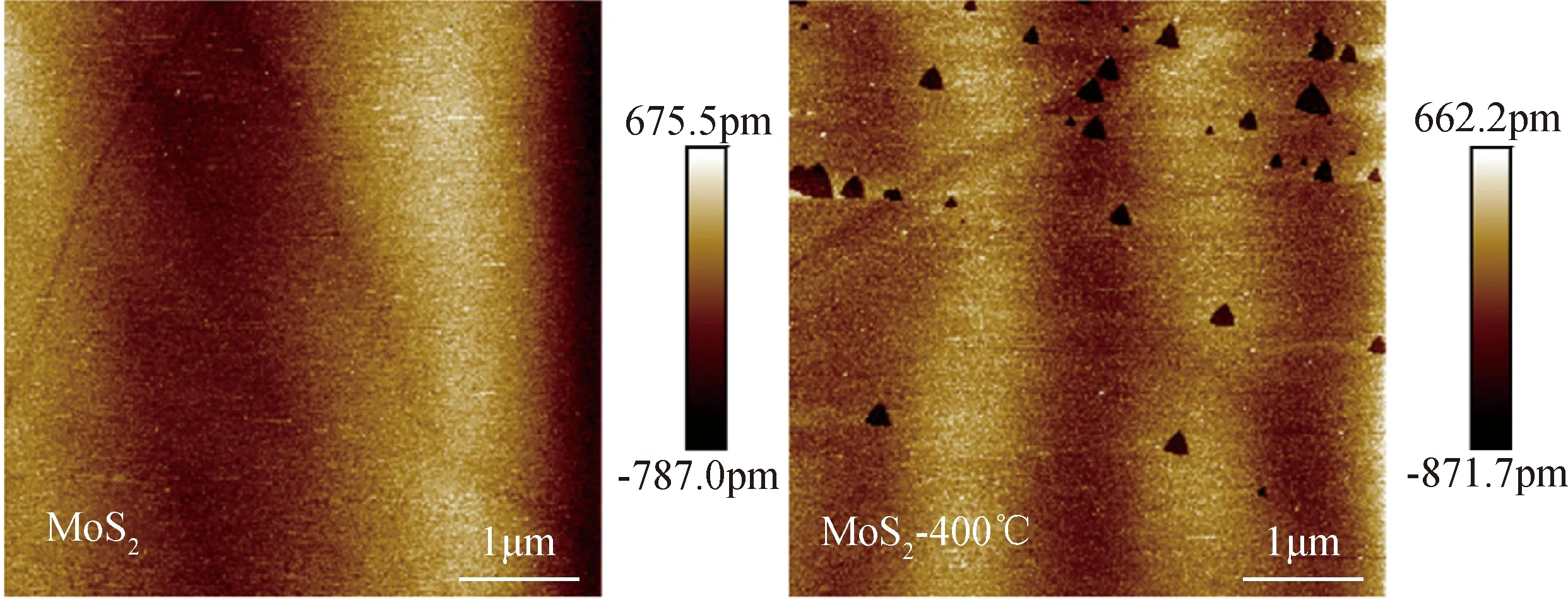
圖4 熱處理前后輝鉬礦AFM照片Fig.4 AFM images of molybdenite before and after thermal modification
2.2 熱處理輝鉬礦光催化原位還原銀離子性能分析
熱處理前后的輝鉬礦對Ag+的還原動力學如圖5(a)所示。為更好地了解其過程和速率,對相關數據進行擬合分析。從圖中可以看出,兩種材料對Ag+的還原效果呈現相同的趨勢,均由快轉慢并逐漸趨于平衡,平衡的出現可能是因為活性位點被占滿。當達到還原平衡時,輝鉬礦的還原量為17 mg/g,而經過熱處理的輝鉬礦還原量為23 mg/g,因此經過熱處理的輝鉬礦還原Ag+的性能要遠強于輝鉬礦。
溶液pH值對熱處理輝鉬礦還原Ag+的過程有重要的影響,影響熱處理輝鉬礦的表面電荷強度。如圖5(b)所示,研究了pH值從1.5至4對熱處理輝鉬礦原位還原Ag+的影響。從圖中可以看到,隨著pH值從1.5增加到3.0,熱處理輝鉬礦對Ag+的還原量逐漸增加,在pH值為3.0時,達到最大,隨后逐漸下降。因此在溶液pH值為3.0時,熱處理輝鉬礦對Ag+的還原效果最好,在后續的試驗研究中,均調節溶液pH值為3.0。
圖5(c)為銀溶液的初始濃度對熱處理輝鉬礦原位還原Ag+的影響結果。從圖中可以看出,熱處理輝鉬礦對Ag+的原位還原能力隨著溶液初始濃度的增加,先迅速上升后趨于平緩,這可能是因為開始時熱處理輝鉬礦界面活性位點豐富,Ag+迅速靠近輝鉬礦表面,并被原位還原成Ag單質,隨著反應的進行,材料表面活性位點被占據,對Ag+的還原能力逐漸飽和。
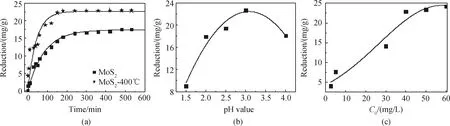
圖5 熱處理前后的輝鉬礦還原Ag+動力學行為(a),溶液pH值(b),及Ag+初始濃度(c)對熱處理輝鉬礦還原Ag+的影響Fig.5 Reduction Ag+ dynamic performance on molybdenite before and after thermal modification(a), solution of pH (b) and initial Ag+ concentration (c) on Ag+ reduction using thermally modified molybdenite, respectively
為了證明Ag元素的存在狀態,采用XPS對吸附Ag+后的熱處理輝鉬礦進行分析,Ag元素的窄譜圖如圖6所示。從擬合結果可知,兩個明顯的特征峰位于結合能為368.6 eV和374.65 eV處,分別對應Ag 3d5/2和Ag 3d3/2的峰,根據前人研究結果可知[27],這是Ag單質的峰,即輝鉬礦表面Ag以單質的形式存在,說明熱處理輝鉬礦可以通過原位還原來回收硝酸銀溶液中的Ag+。
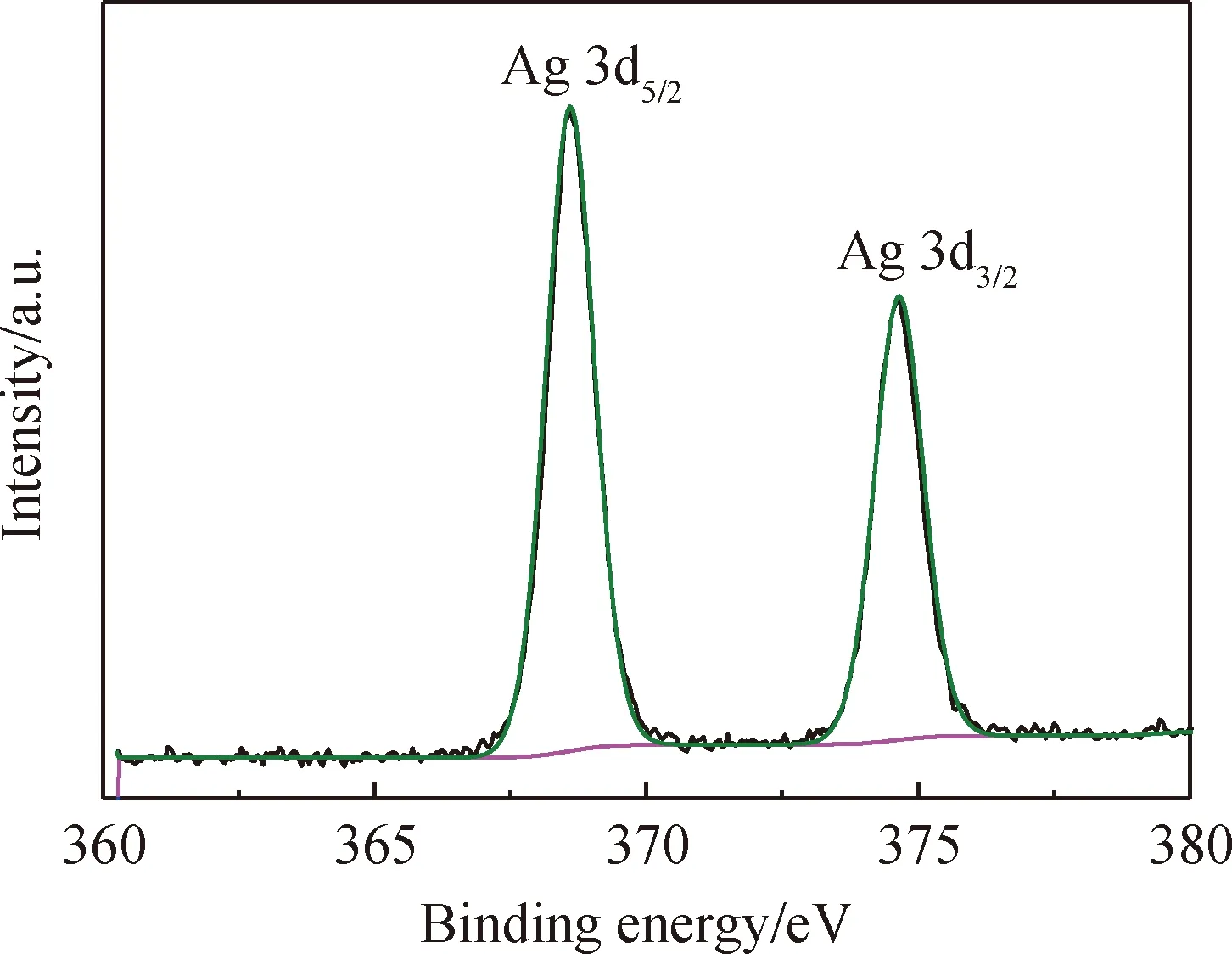
圖6 熱處理輝鉬礦還原Ag+后的Ag 3d的XPS窄譜Fig.6 XPS spectra for Ag 3d of thermally modified molybdenite after reduction
圖7為熱處理輝鉬礦還原銀后的SEM-EDS結果。從圖(a)中可以看出,熱處理輝鉬礦為片狀結構,表面較為粗糙。而從圖(b)、(c)、(d)中可以看出樣品中主要含有Mo、S、Ag等元素。由(d)圖測試結果可知,Ag元素均勻的分布在熱處理輝鉬礦表面,而右下角處可歸為Ag單質的存在。
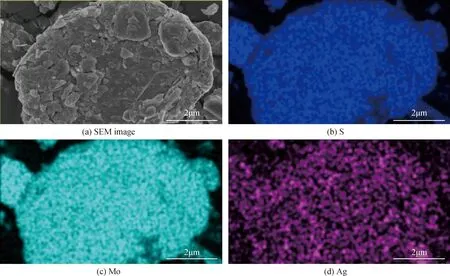
圖7 熱處理輝鉬礦還原Ag+后的SEM-EDS照片Fig.7 SEM-EDS images of thermally modified molybdenite after reduction
2.3 機理解釋
為探究熱處理對輝鉬礦還原銀的促進機理,對熱處理前后輝鉬礦進行了紫外可見吸收光測試,結果如圖8所示。由圖可知,熱處理前后,輝鉬礦展現的吸收峰數量及位置均未發生變化,再次證明熱處理并未改變輝鉬礦的結構。熱處理后各吸收峰的強度增加,說明熱處理后輝鉬礦的光吸收能力提高,從而增強其原位光還原Ag+效果。
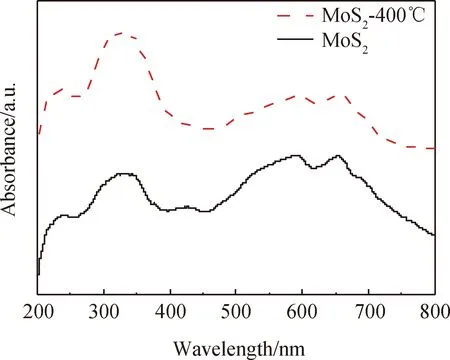
圖8 輝鉬礦熱處理前后的紫外可見譜Fig.8 UV-vis spectra of molybdenite before and after thermal modification
3 結 論
(1)熱處理對輝鉬礦的晶體結構影響較小。
(2)熱處理可顯著提高輝鉬礦對Ag+的原位還原性能,由原礦的17 mg/g提升到熱處理后的23 mg/g。
(3)在Ag+初始濃度為50 mg/L及溶液pH值為3.0的條件下,熱處理輝鉬礦原位還原Ag+效果最好。
(4)熱處理對輝鉬礦原位還原Ag+效果的提升來自于其對可見光吸收強度變高及其活性位點變多。
