HPLC和UV法對比測定蚊不叮霜中VB1的含量
2020-10-21 01:40:18朱家航朱金芳梁嘉培巴餓江努哈依甫
新疆農業科學 2020年10期
朱家航,李 艷,朱金芳,梁嘉培,巴餓江·努哈依甫
(1.新疆農業大學,烏魯木齊 830052;2.新疆銀朵蘭維藥股份有限公司,烏魯木齊 830000)
0 引 言
【研究意義】維生素B1又稱硫胺素,分子式為C12H17ClN4OS·HCl,屬于水溶性維生素B族中的一種,是維持機體正常代謝及功能不可或缺的一類低分子化合物,對神經系統起到保護作用。其在酸性溶液中很穩定,在堿性溶液中不穩定,易被氧化而分解變質,光、熱對其影響較大,故應置于遮光、涼處保存[1-2]。建立UV和HPLC法測定蚊不叮霜中維生素B1含量的方法,對蚊不叮霜中維生素B1含量測定有實際意義。【前人研究進展】由于分子中存在共軛雙鍵結構,因此,在紫外區有吸收峰,最大吸收波長為246 nm,對應的百分吸收系數為421。李艷霞等[3]研究發現在NaOH介質中,維生素B1可將 K3[Fe(CN)4]定量還原為K4[Fe(CN)4]與 Fe3+發生反應生成可溶性普魯士藍,由此建立了鐵氰化鉀分光光度法間接測定維生素B1的含量。王瑞勇等[4]基于在酸性條件下,檸檬酸鈉將氯金酸還原成納米金后可與維生素B1發生相互作用,納米金發生凝聚,其最大吸收峰產生了紅移,建立了比色測定法測定維生素B1的含量,成功定量分析了片劑和注射液中的維生素B1。李丹鳳[5]采用 HLPC 法同時測定了三維葡磷鈣咀嚼片中VB1和VB2的含量。呂惠卿[6]建立了高效液相色譜-熒光檢測法測定了復合維B片中VB1的含量,簡便、快速、準確。韓宇等[7]以二極列管檢測器(DAD)進行檢測同時測定了復方氨基丁酸維E膠囊中VB1和鹽酸的含量。維生素B1的含量測定方法主要有UV法[8]、HPLC法[9-14]、核磁共振法[15]、熒光分析法[16]、化學發光法[17]及毛細管電泳法[18]等。《中國藥典》2015版(二部)[19]中采用UV法測定其測定維生素B1片劑及注射液中維生素B1的含量,簡單、可靠。采用HPLC法檢查其有關物質。【本研究切入點】近年來,針對于維生素B1的測定多集中于UV法和HPLC 法,就2種方法測定蚊不叮霜中維生素B1的含量進行對比,【擬解決的關鍵問題】研究對比UV法和HPLC法測定蚊不叮霜中維生素B1的含量,為蚊不叮霜中維生素B1含量測定方法的確定提供依據。
1 材料與方法
1.1 材 料
1.1.1 儀 器
高效液相色譜儀(LC-20,日本島津公司);紫外分光光度儀(UV2550,日本島津公司);紫外-可見分光光度計(T6新世紀,北京普析通用儀器有限責任公司);電子天平(AL204-IC,梅特勒-托利多儀器(上海)有限公司);pH計(pHS-3C,上海儀電科學儀器股份有限公司);電熱恒溫水浴鍋(W201,上海申生科技有限公司);高速冷凍離心機(SF-GL-16A,上海非恰爾分析儀器有限公司);真空抽濾泵(VP50,武漢集思儀器設備有限公司)。
1.1.2 試 劑
蚊不叮霜(自制,規格10 g : 80 mg,批號20181225-1,20181225-2,20181225-3);維生素B1對照品(購于中國食品藥品檢定研究院,純度97.9%);甲醇、乙腈、庚烷磺酸鈉、磷酸、三乙胺為色譜純,鹽酸為分析純,水為超純水。
1.2 方 法
1.2.1 UV法測定VB1含量
1.2.1.1 溶液的配制
(1)供試品溶液
精密稱取蚊不叮霜1 g(約相當于維生素B18 mg)于50 mL碘量瓶中,加入鹽酸溶液約40 mL,70℃水浴加熱并不斷振搖10 min后,再置冰水浴(0℃)中冷卻20 min,過濾至100 mL容量瓶中,按上述步驟用鹽酸溶液重復提取膏體部分2次,每次約30 mL,將3次提取液合并至100 mL容量瓶中定容,搖勻。精密吸取1 mL,置于10 mL容量瓶中,用鹽酸溶液稀釋至刻度,即得。
(2)陰性對照溶液
精密稱取不含維生素B1的蚊不叮霜陰性對照樣品1 g,按“1.2.1.1”項下,供試品溶液的配制方法制備。
(3)對照品儲備液
精密稱取適量干燥至恒重的維生素B1對照品25 mg,置于100 mL容量瓶中,加入鹽酸溶液約70 mL搖勻溶解維生素B1,加入鹽酸溶液稀釋至刻度,搖勻,制成0.25 mg/mL的對照品儲備液,并將其放入冰箱中冷藏。
(4)對照品溶液
精密吸取對照品儲備液5 mL,置于100 mL容量瓶中,用鹽酸溶液稀釋至刻度,搖勻,制成12.45 μg/mL的對照品溶液。
(5)鹽酸溶液
鹽酸溶液(0.1 mol/L):量取濃鹽酸溶液9 mL,用水稀釋并定容至1 000 mL,搖勻。
1.2.1.2 檢測波長
取適量的對照品溶液和陰性對照溶液,在200~800 nm波長范圍內進行波長掃描。
1.2.1.3 線性關系
精密吸取對照品儲備液0.30、0.35、0.40、0.45、0.50、0.55、0.60 mL,分別用鹽酸溶液配置成7.47、8.72、9.96、11.21、12.45、13.70、14.94 μg/mL系列濃度后,測定。
1.2.1.4 精密度
取對照品溶液適量,以溶劑為空白,在246 nm處測定吸光度值,連續測定6次,記錄吸光度值。
1.2.1.5 重復性
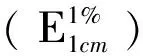
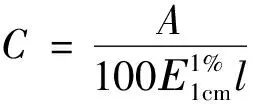
C—供試品溶液的濃度(g/mL);A—吸光度值;E:吸收系數;l—比色皿液層厚度(cm);D—稀釋倍數;W—供試品的取樣量(g)。
1.2.1.6 加標回收率
分別精密稱取6份已知含量的供試品(批號20181225-1),每份約0.5 g,分別精密加入維生素B1對照品儲備溶液17 mL,按“1.2.1.1”項下,供試品溶液配制方法配制,測定。
1.2.1.7 穩定性
取同一供試品溶液,室溫條件下放置,分別于0、5、10、20、30 min測定。
1.2.2 HPLC法測定VB1含量
1.2.2.1 溶液的制備
(1)供試品溶液
精密稱取蚊不叮霜1 g(約相當于維生素B18 mg)于50 mL碘量瓶中,加入鹽酸溶液約40 mL,水浴加熱并不斷振搖10 min后,再置冰水浴中冷卻20 min,過濾至100 mL容量瓶中,按上述步驟用鹽酸溶液重復提取膏體部分2次,每次約30 mL,將3次提取液合并至100 mL容量瓶中定容,搖勻。精密吸取1 mL,置于10 mL容量瓶中,用流動相稀釋至刻度。
(2)對照品儲備液
精密稱取適量干燥至恒重的維生素B1對照品10.1 mg,置于25 mL容量瓶中,用流動相稀釋至刻度,搖勻,配制成0.404 mg/mL的儲備液。
(3)對照品溶液
精密吸取對照品儲備液1 mL,置于50 mL容量瓶中,用流動相稀釋至刻度,搖勻,制成8.08 μg/mL的對照品溶液。
1.2.2.2 檢測波長
取適量的對照品溶液,在200~800 nm波長范圍內進行掃描。
1.2.2.3 色譜條件
色譜柱:Diamonsil C18柱(150×4.6 mm,5 μm);
流動相:甲醇-乙腈-0.02 mol/L庚烷磺酸鈉溶液(含1%三乙胺,用磷酸調pH值至5.5)=9∶9∶82;
流速:1.0 mL/min;
檢測波長:267 nm;
進樣量:20 μL;
柱溫:室溫;
采用外標法峰面積定量。
1.2.2.4 系統適用性
在上述色譜條件下,取空白溶劑、維生素B1對照品溶液、供試品溶液和陰性對照溶液進樣。
1.2.2.5 線性關系
精密吸取對照品儲備液0.6、0.8、1.0、1.2、1.4 mL,用流動相分別配置成4.85、6.46 μg/mL、8.08、9.70、11.31 μg/mL系列濃度測定。
1.2.2.6 精密度
取對照品溶液適量,按上述色譜條件連續測定6次,記錄峰面積。
1.2.2.7 重復性
分別精密稱取同一批號的蚊不叮霜(批號20181225-1)6份,按“1.2.2.1”項下,供試品溶液制備方法配制,按上述色譜條件測定維生素B1的峰面積,按外標一點法計算含量。
1.2.2.8 加標回收率
精密稱取6份已知含量的供試品(批號20181215-1),每份約0.5 g,分別精密加入維生素B1對照品儲備液10 mL,按“1.2.2.1”項下配制方法,按上述色譜條件測定。
1.2.2.9 穩定性
取同一供試品溶液,于室溫下放置0、2、4、6、8、12 h,按上述色譜條件測定。
1.2.3 三批樣品含量
三批樣品(批號20181225-1、20181225-2、20181225-3)分別進行HPLC法和UV法的測定。
2 結果與分析
2.1 UV法測定維生素B1含量
2.1.1 檢測波長的選擇
研究表明,在246 nm處有最大吸收,維生素B1陰性對照溶液200~800 nm結果為在246 nm處無吸收,表明在246 nm處測定維生素B1有最大吸收且其他成分無干擾。選定246 nm為測定波長。圖1,圖2
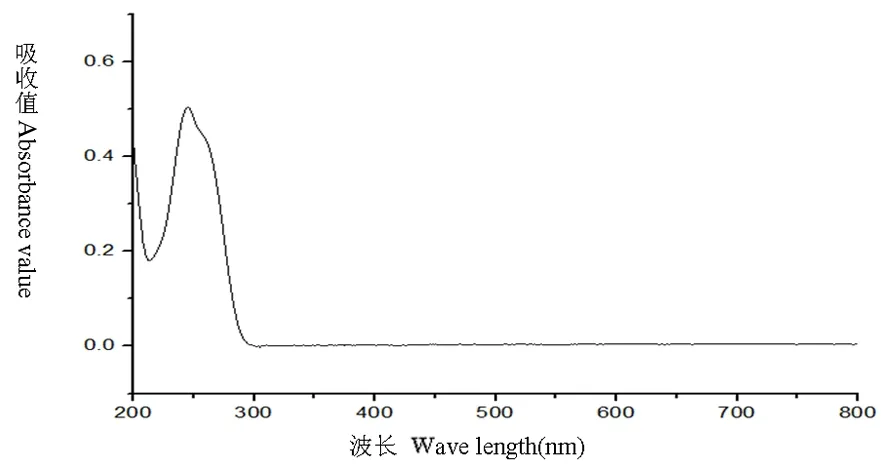
圖1 維生素B1對照品溶液全波
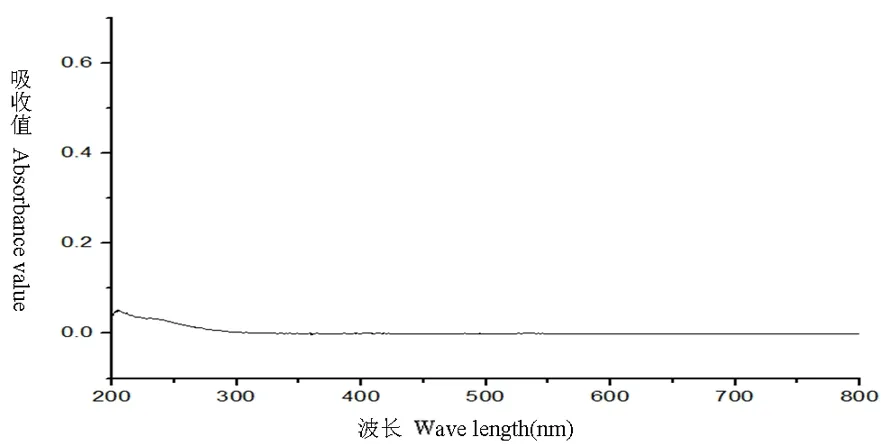
圖2 陰性對照溶液全波
2.1.2 線性關系
以濃度(C)為橫坐標,吸光度值(A)為縱坐標,進行線性回歸。維生素B1對照品線性回歸方程為A=0.045 9C-0.063 4,r=0.999 3。說明維生素B1在 7.47~14.94 μg/mL范圍內,線性關系良好。圖3
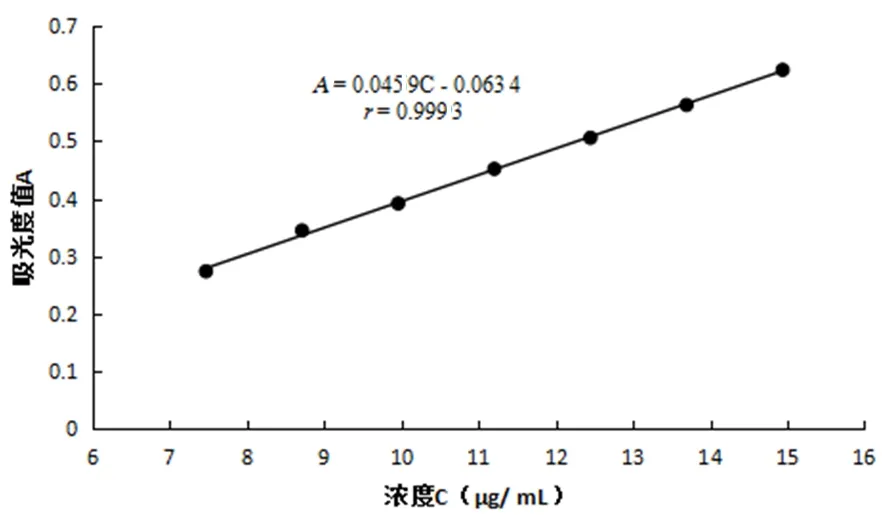
圖3 維生素B1標準曲線
2.1.3 精密度
維生素B1含量RSD=0.30%,儀器精密度良好。表1
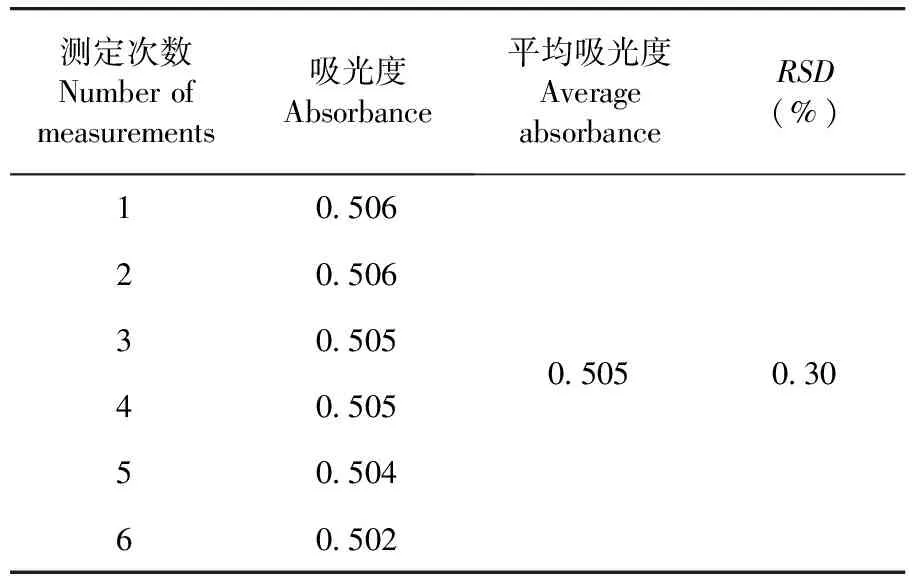
表1 精密度(n=6)
2.1.4 重復性試驗
通過計算得出平均含量為8.72 mg/g,RSD=1.04%(n=6),表明該方法重復性較好。表2
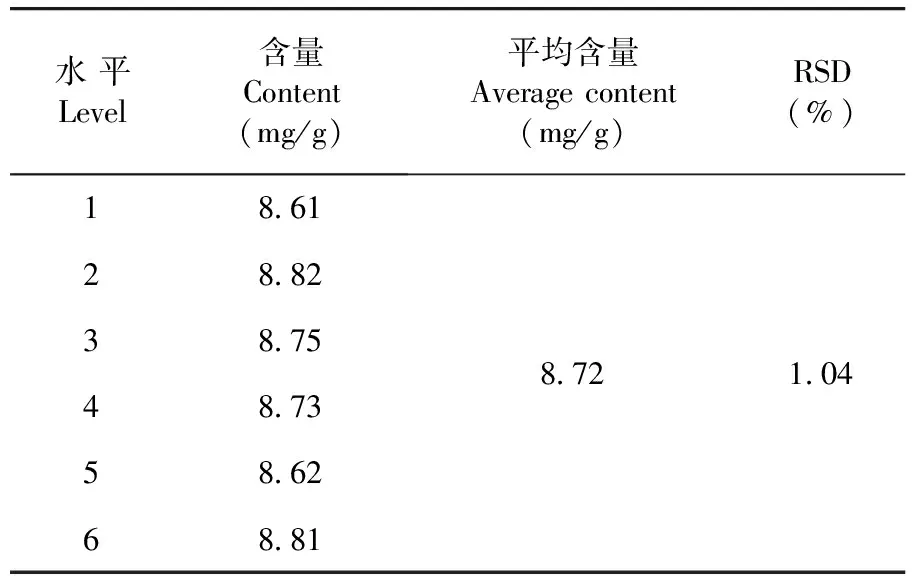
表2 重復性
2.1.5 加標回收率試驗
測得平均回收率為105.13%,RSD=1.72%,表明回收率良好,準確度高。表3
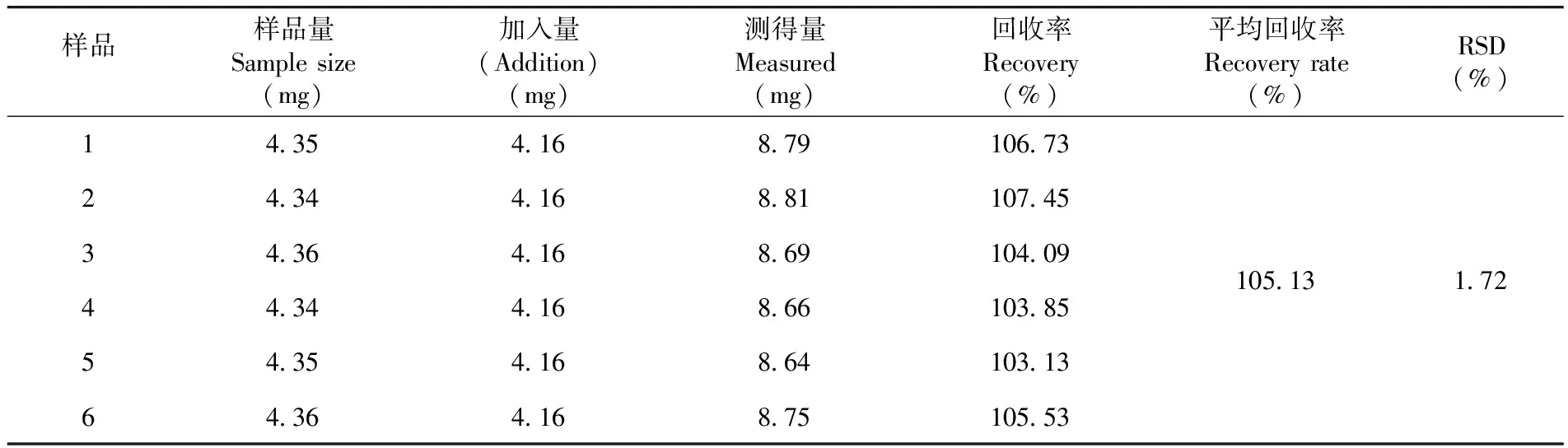
表3 加標回收率
2.1.6 穩定性
穩定性試驗結果RSD=0.91%,供試品溶液在室溫放置30 min穩定性良好。表4
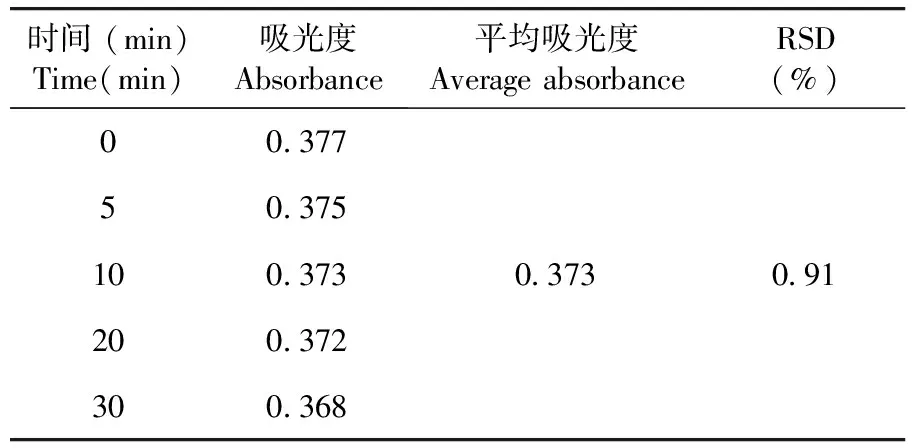
表4 穩定性
2.2 HPLC法測定維生素B1含量
2.2.1 檢測波長的選擇
研究表明,在267 nm處有最大吸收,表明在267 nm處測定維生素B1有最大吸收。選定267 nm為測定波長。圖4
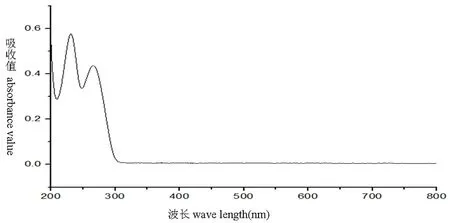
圖4 維生素B1對照品溶液全波
2.2.2 線性關系
樣品中維生素B1與其他組分能夠分離,理論塔板數(n>3 000),該方法專屬性好,其他成分對測定無干擾。圖5~8
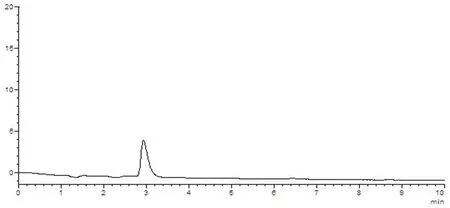
圖5 溶劑空白HPLC圖譜
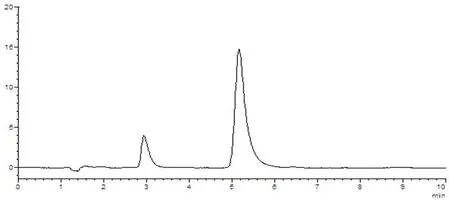
圖6 維生素B1對照品溶液HPLC圖譜
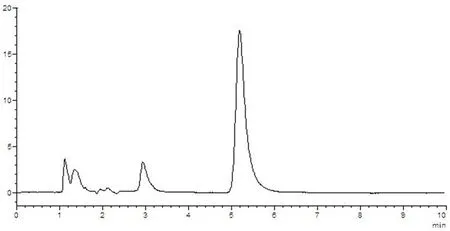
圖7 供試品溶液HPLC圖譜
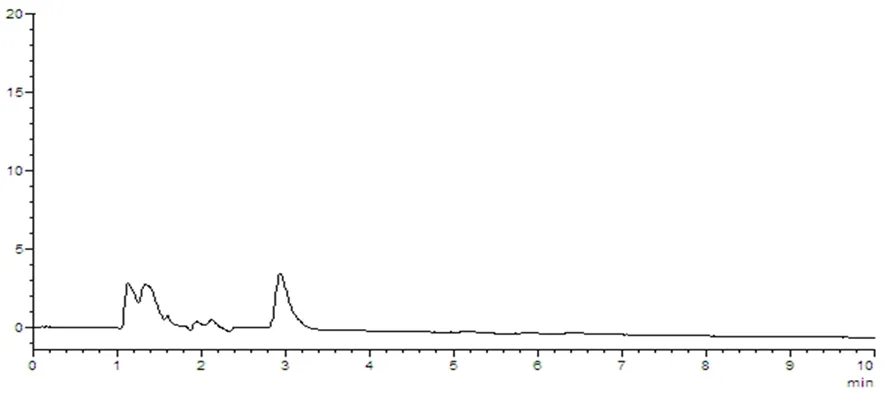
圖8 陰性對照樣品溶液HPLC圖譜
2.2.3 線性關系考察
以濃度(C)為橫坐標,峰面積(Are)為縱坐標,進行線性回歸,回歸方程為A=31 146C+4 562,r=0.999 9。表明維生素B1在4.85~11.31 μg/mL線性關系良好。圖9
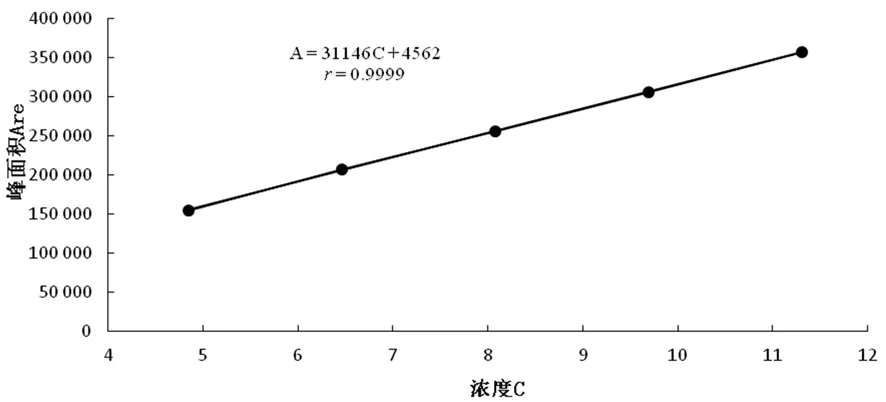
圖9 維生素B1標準曲線
2.2.4 精密度
維生素B1峰面積RSD=0.28%,儀器精密度良好。表5
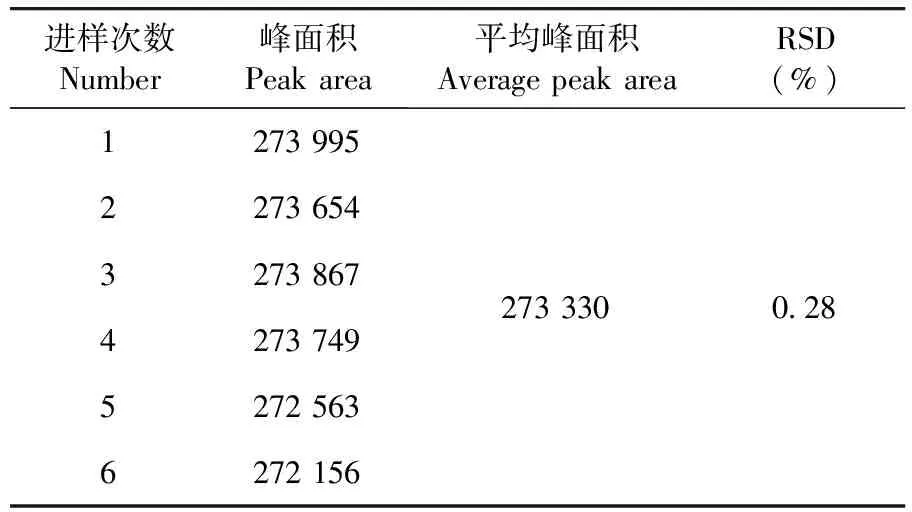
表5 精密度實驗結果
2.2.5 重復性
測得平均含量為8.39 mg/g,RSD=0.90%(n=6),該法重復性良好。表6
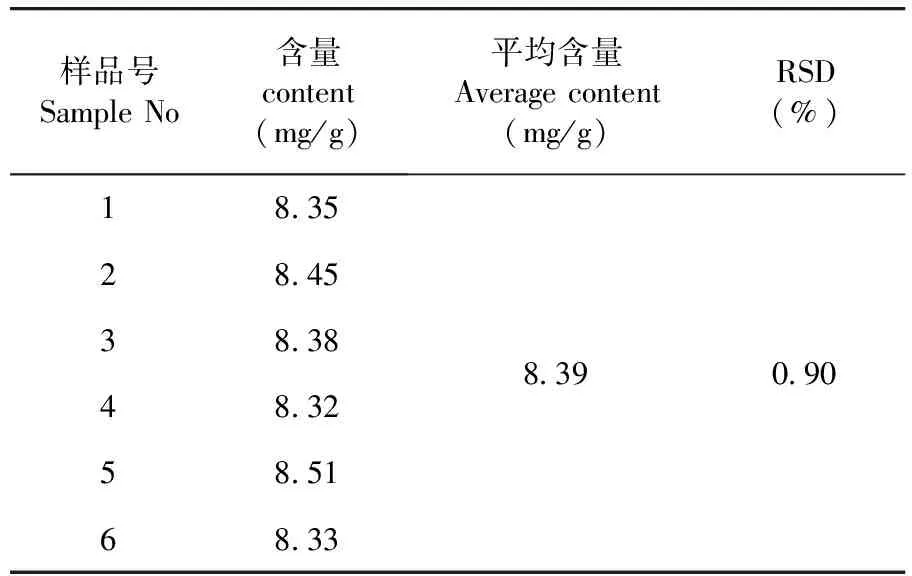
表6 重復性試驗結果
2.2.6 加標回收率
測得平均回收率為101.77%,RSD=1.93%,回收率良好,準確度高。表7
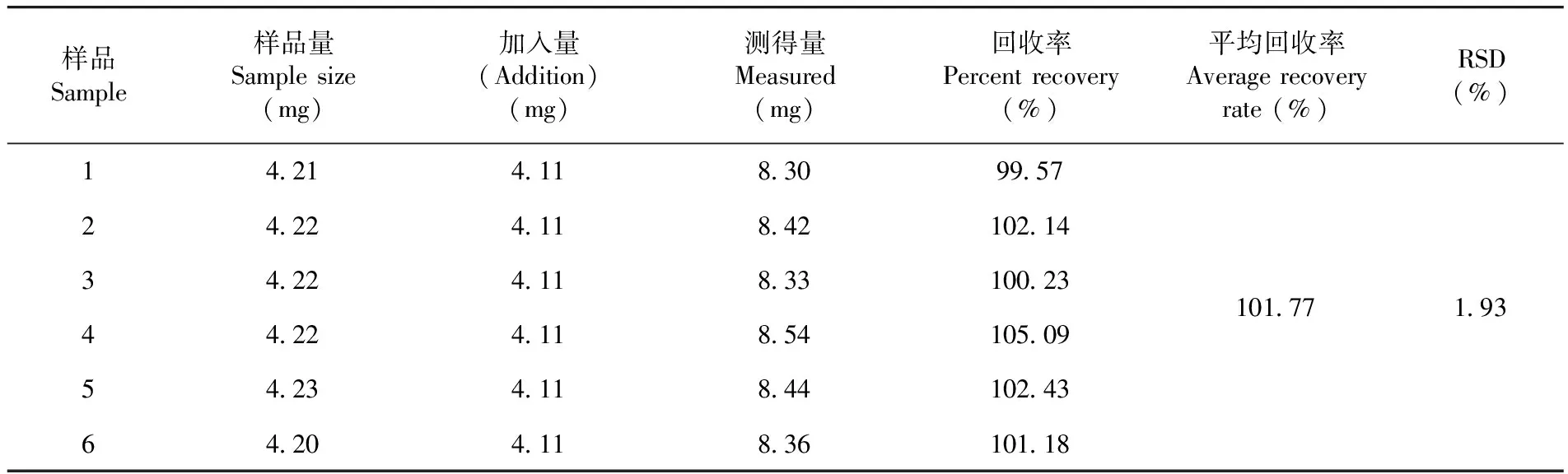
表7 加標回收率試驗結果(n=6)
2.2.7 穩定性
穩定性實驗結果RSD=0.39%,供試品溶液在室溫放置12 h穩定性良好。表8
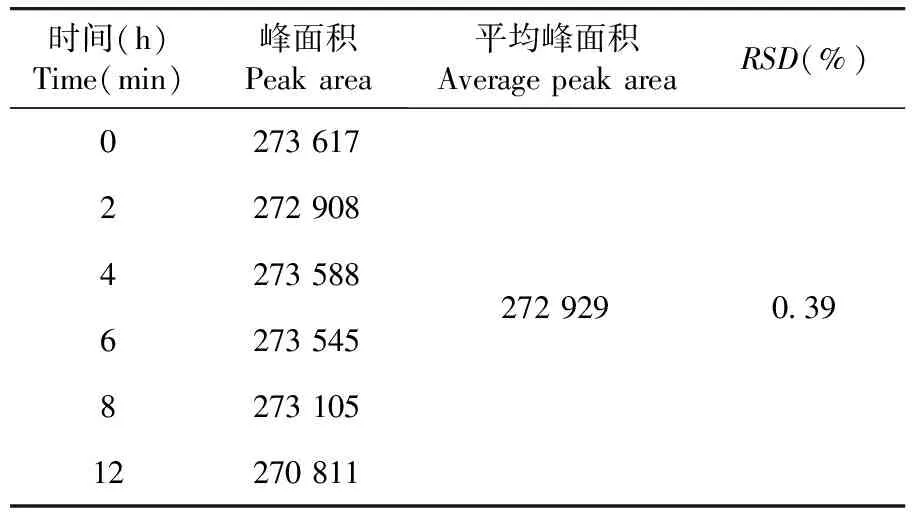
表8 穩定性
2.3 三批樣品含量
HPLC法測得三批樣品平均含量分別為8.39 mg/g、8.39 mg/g、8.45 mg/g;UV法測得三批樣品平均含量分別為8.73 mg/g、8.70 mg/g、8.77 mg/g。與蚊不叮霜的標示量(8 mg/g)相比,雖然UV法和HPLC法測得結果均在合格范圍內(標示量的90%~110%,即7.2~8.8 mg/g),但UV法測得的含量偏高,準確度較HPLC法差。表9
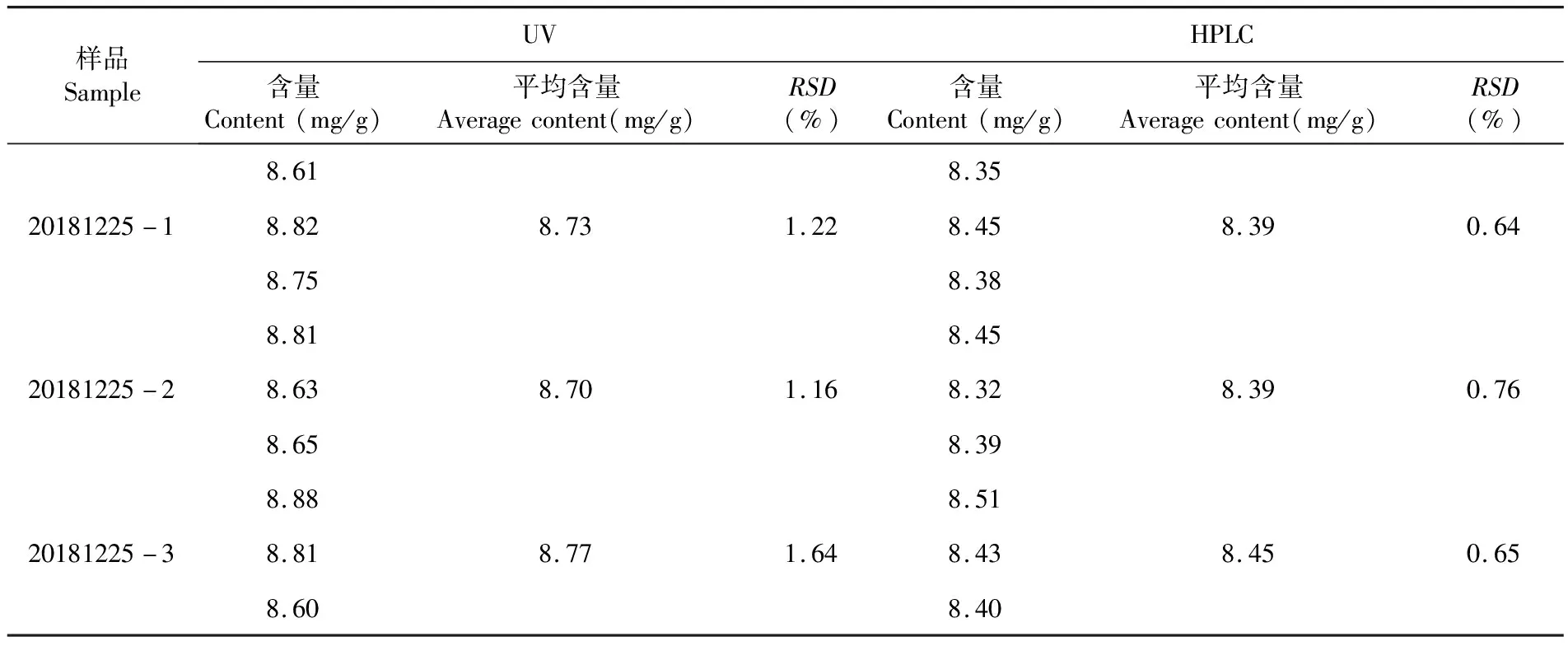
表9 三批樣品含量
3 討 論
參考《中國藥典》2015版(二部)[19]中維生素B1有關物質檢查的HPLC條件,將HPLC流動性確定為甲醇-乙腈-0.02 mol/L庚烷磺酸鈉溶液(含1%三乙胺,用磷酸調pH值至5.5)。經測定,在此流動相條件下,色譜峰峰形良好,柱效、分離度均達到要求,故選定該流動相為本次實驗所用流動相。
UV法的優點為操作簡單、用時少、對儀器設備的要求不高[1,8],其缺點是易受樣品中輔料等其他物質的干擾,使測得結果偏大;而HPLC法具有較好的分離效果,能夠排除其他成分的干擾,測定準確性高[5,7,10]。研究通過對比UV法和HPLC法在測定蚊不叮霜中精密度、重復性、回收率及測定樣品含量的準確度,最終確定HPLC測定蚊不叮霜中維生素B1含量的方法較優。
4 結 論
UV法及HPLC法的精密度、重復性均較好,UV法測得三批平均含量分別為8.73、8.70和8.77 mg/g,平均回收率105.13%;HPLC法測得平均含量分別為8.39、8.39和8.45 mg/g,平均回收率為101.77%,對比得出UV法較HPLC法測得三批平均含量分及回收率均偏高,故測定準確度不如HPLC法,且UV法穩定性較差,只能在室溫下30 min穩定,HPLC法穩定性可以在室溫12 h穩定,確定專屬性和穩定性均較高HPLC法測定蚊不叮霜中維生素B1的含量。
