硫化物和硫/磷化合物的添加方式對石腦油熱裂解結焦影響的研究
2020-11-18 01:56:20王志遠丁旭東王博研邢志宏
化工學報 2020年11期
王志遠,丁旭東,王博研,邢志宏
(上海理工大學能源與動力工程學院,上海市動力工程多相流動與傳熱重點實驗室,上海200093)
引 言
裂解爐管內壁面焦炭的沉積不僅導致傳熱效率降低,系統能耗提高,裝置處理量減少,產品收率降低,而且造成管壁滲炭,爐管的使用壽命被縮短。近三十年來,硫化物對于反應器壁積炭、催化劑失活的影響等方面的研究一直是人們關注的焦點[1-3]。在烴類熱裂解過程中,原料中的含硫組分可以與爐管表面具有催化活性的金屬形成硫化物,或以吸附態硫的形式鈍化爐管,從而抑制爐管材料中鐵和鎳元素的催化脫氫作用,降低催化結焦速率[4]。硫化物對于熱裂解結焦過程具有復雜的影響,主要取決于硫化物自身性質和添加方式。最近,比利時Gent大學油品實驗室的研究還表明[1],硫化物對于熱裂解結焦行為的影響還取決于合金的預處理方式。
實驗室多采用硫醇等簡單硫化物作為抑制結焦手段,使用方法為將硫化物直接加入至裂解原料或裂解前對爐管進行預處理,但抑制效果一般[2-4]。在烴類熱裂解過程中,考慮到操作安全性、裂解條件下熱力學和動力學的統一性、對下游操作單元影響及經濟性等因素,磷化物(磷酸酯、磷酸鹽化合物等)也被作為抑制劑使用[5-6]。研究表明[5],磷化物主要通過屏蔽催化活性金屬來抑制管壁的催化作用,降低結焦速率。Bao 等[7]在考察硫磷化合物對MnCr2O4氧化物表面結焦行為影響時還發現,磷化物自由基改變了結焦層形態,減輕和防止金屬表面與炭粒之間、炭粒與炭粒之間的黏附,使焦炭松散易除。因此,為了充分發揮硫化物抑制催化活性中心的功效,減小其對爐管金屬腐蝕的副作用,可以采用硫化物與磷化物聯合操作的方式。目前,國內外研究多集中于單一物系化合物下的結焦行為考察,而關于硫化物與相關化合物聯合操作下烴類熱裂解結焦的研究較少,尤其是關于硫/磷化合物添加方式方面的實驗還未見報道。本文較為系統地考察了較短裂解時間內硫化物與硫/磷化合物的添加方式對石腦油熱裂解結焦行為的影響,并通過結焦層形貌與結構的表征結果,對不同添加方式的抗結焦機理進行探討和比較。
1 實驗材料和方法
1.1 材料
合金試樣:HP40合金(25Cr-35Ni),線切割為尺寸20 mm×10 mm×2 mm 片狀試樣;硫化物:二甲基二硫[(CH3)2S2,DMDS,等級RG,純度99%+,Adamas公司]、噻吩(C4H4S,等級RG, 純度99%, Adamas 公司)、苯并噻吩(C8H6S, 等級RG, 純度99%,Adamas公司);磷化物:亞磷酸三乙酯(C6H15O3P,TEP,等級RG, 純度99%, Adamas 公司)、亞磷酸三苯酯(C18H15O3P,TPPI,等 級RG, 純 度99%, Adamas 公司);溶劑:乙醇(C2H5OH,等級AR,純度>99.7%,Greagent 公司)、乙二醇(C2H6O2,等級AR,99.5%,Adamas 公司);裂解原料:輕石腦油(餾程:室溫~120℃,硫質量濃度<20 μg/g)。
1.2 熱裂解結焦評價實驗
1.2.1 合金試樣的預氧化處理 采用180#~400#水磨砂紙對式樣打磨,并于丙酮和乙醇中超聲清洗。將清洗后的試樣置于管式爐,進行程序控溫氧化處理,過程如下:①氮氣氣氛中,以5℃/min速率升溫至200℃,保溫0.5 h,以脫除式樣表面殘余水分;②空氣氣氛中,以10℃/min 速率繼續升溫至800℃,保溫10 h,空氣流量為700 ml/min;③自然冷卻至室溫后,使用去離子水對試樣進行清洗。
1.2.2 合金試樣的預硫化及硫/磷化合物預處理 裂解實驗之前,可利用硫化物或硫/磷化合物對式樣進行預處理。預硫化處理過程如下:①氮氣氣氛下,反應器以10℃/min速率升溫至800℃;②進行模擬工業裂解爐進料前的清焦操作,反應器在空氣氣氛中進一步升溫至850℃,保持空氣流量為500 ml/min,該過程持續約20 min;③將二甲基二硫加入乙醇與乙二醇配比成的溶液中(3 ml,Vethanol∶Vglycol=1∶2),混合物溶解于去離子水后,利用磁力攪拌器繼續攪拌20 min;④高溫蒸汽氣氛下,反應器降溫至830℃,混合物由計量泵輸運至反應器,預硫化過程為2 h。硫/磷化合物預處理過程與此相同。
1.2.3 裂解實驗 熱裂解結焦實驗在自制熱裂解結焦評價裝置中進行,采用輕石腦油作為裂解原料,水蒸氣作稀釋劑[7-9]。氮氣氣氛下,反應器首先以10℃/min速率升溫至800℃。隨后,氣路由氮氣轉換至空氣,進行模擬裂解爐進料前清焦操作,反應器進一步升溫。當溫度達到850℃,關閉氣路,裂解原料與去離子水由計量泵輸送進入汽化爐,裂解反應開始。當對硫化物及硫/磷化合物結焦性能進行評價時,將化合物直接注入石腦油原料,使用玻璃棒劇烈攪拌,再用磁力攪拌器攪拌20 min。分別稱量試驗前后的試樣質量,兩次質量之差即為試樣結焦量,對3 次試驗結焦量取平均值。裂解結焦評價裝置可重復性已有論述,此處不再贅述[7-9]。
相關實驗條件和實驗方案見表1和表2。

表1 實驗參數Table 1 Experimental conditions for each experimental step
1.3 分析儀器
采用場發射電子顯微鏡(FE-SEM)觀察焦炭的微觀形貌;采用激光拉曼光譜(Raman)分析試樣表面氧化層組成和焦炭化學結構,氬離子激光器,波長514.5 nm,為防止新氧化物的生成和焦炭的燒蝕,激光能量范圍0.07~2.00 mW;采用X射線衍射譜儀(XRD)表征試樣氧化物成分;采用X 射線光電子能譜儀分析焦層表面的官能團含量,單色器Al 靶(1486.6 eV),以C 1s(284.8 eV)峰為內標校正,窄掃通過能為40 eV,步長為0.5 eV,寬掃通過能為160 eV,步長為1 eV;采用德國Elementar Vario EL Ⅲ元素分析儀考察焦炭中氫碳比。

表2 實驗方案Table 2 Experimental plans for each experimental step
2 結果與討論
2.1 HP40合金試樣表面氧化層的成分和結構
圖1 為不同預處理條件下HP40 合金試樣氧化層的Raman 光譜。由于所選用硫/磷化合物具有疏水性,因此,本文采用乙醇/乙二醇混合物作為預處理試劑的溶劑,并對其使用效果進行考察。由圖1(a)可知,當乙醇/乙二醇混合溶液體積過高時,氧化試樣的Raman 譜圖出現了典型的碳的特征峰。這是由于醇類在高溫條件下發生了裂解,并導致了焦炭的沉積[10]。當醇類體積降低至3 ml 后,譜圖中的特征峰消失[圖1(b)],表明該條件下焦炭與水蒸氣汽化速率大于醇類裂解結焦速率。由圖1(b)可知,合金試樣經過高溫預硫化處理后,表面主要生成了由Fe、Cr 元素組成的氧化物。其中,530 cm-1位置的弱峰對應于Cr2O3,300 cm-1、560 cm-1位置的峰位對應于Fe3O4,645 cm-1、1420 cm-1位置的峰位對應于γ-Fe2O3,665~680 cm-1的 峰位 對應 于Fe 的 氧化 物(Fe3O4和γ-Fe2O3)[11-13]。圖1(c)表明,不同預處理條件下,HP40合金表面氧化物成分類似。
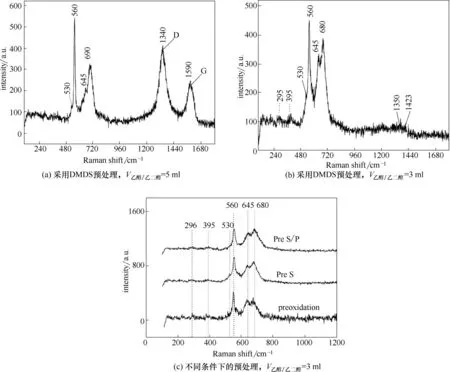
圖1 不同預處理條件下HP40合金表面氧化層的Raman光譜Fig.1 Raman spectra of the oxide films on HP40 alloys
圖2 為不同預處理條件下HP40 合金試樣氧化層的XRD 譜圖,為了詳細分析預處理過程對氧化層組成的影響,對譜圖局部進行放大觀察。由圖2(a)可知,氧化層中合金Cr2O3峰和Fe3O4峰較為明顯,這與Raman 光譜的表征結果一致。而經過硫化物以及硫磷化合物處理后,氧化層中還生成了少量的NiFe2O4、MnFe2O4和Fe-2CrO4等含Fe 元素的氧化物結構,表明氧化層中Fe含量升高。根特大學利用熱力學軟件EkvicalCal 對HP40 合金表面氧化層組分的計算結果發現,與空氣氧化處理相比,預硫化使合金表面Fe、Ni含量升高,Cr含量降低[1,14]。
圖3 為不同預處理條件下HP40 合金試樣氧化層的SEM 照片和EDS分析結果。由SEM 圖可知,試樣表面經過硫化物或硫/磷化合物預處理后,氧化層主要由片狀(blade-like oxides)和粒狀的氧化物結構組成,而空氣氧化試樣表面氧化層主要由粒狀形貌的氧化物組成。金屬氧化物通常呈現粒狀形貌特征,當高溫氧化氣氛中含有水蒸氣時,合金表面氧化物會出現片狀形貌,片狀結構氧化物的生成與金屬沿氧化層中螺旋位錯的快速輸運有關[15]。金屬表面生成的具有一定形狀的氧化物結構對于其表面結焦行為具有復雜影響:一方面,提高了合金表面的粗糙度與活性位數目,使得結焦母體在氧化層表面的停留時間延長,導致結焦量增加[3,14];另一方面,氧化物可以增加硫磷自由基在合金表面的吸附面積,使得氧化層表面催化活性降低[7,9]。AFM 測試結果表明,不同預處理條件下試樣表面氧化層粗糙度(Ra)范圍0.1~0.2 μm,硫化物或硫/磷化合物預處理未造成氧化層粗糙度發生明顯變化。
采用EDS 對不同預處理條件下的氧化層進行分析,不同加速電壓下的結果見表3。能譜電子束滲透深度采用式(1)計算[6,14]:

式中,Wa為元素摩爾質量,g/mol;Z 為原子數;ρ為基底材料密度,g/cm3;E 為加速電壓,kV。根據式(1),當加速電壓為10 kV 時,EDS 的檢測深度約為0.53 μm;而當加速電壓達到15 kV 時,EDS 的檢測深度可達到1.04 μm。由表3 可知,與空氣氧化相比,試樣表面經過硫化物或硫/磷化合物預處理后,在氧化層表面0~1 μm 范圍內,Fe、Ni、Mn 元素含量略有升高,而Cr 元素含量略有下降,這與XRD 的表征結果吻合。不同預處理條件下,氧化層中的Cr、Mn 含量高于合金基體中的含量,而Fe、Ni 含量則低于基體。在高溫氧化氣氛中,Cr、Mn元素在HP40合金中的擴散速率遠高于其他合金組成元素。因此,Cr、Mn可以迅速擴散至金屬表面并形成氧化物[9,15]。
2.2 硫化物的連續添加對熱裂解結焦行為的影響
圖4 為加入不同硫化物后(原料中硫質量分數Csulfur=136 μg/g),HP40 合金氧化試樣表面的焦炭形貌。由圖可見,硫化物的加入明顯改變了焦層的形貌。當裂解時間為1 h,絲狀焦交織布滿空白試樣表面,顯示出明顯的催化結焦特征。由合金試樣氧化層的Raman 表征結果可知(圖1),氧化層表面主要由Fe3O4和γ-Fe2O3等組成,且Fe3O4對應的峰位較強。相比于Fe,Fe3O4具有更強的催化結焦活性[3]。因此,氧化合金試樣表面具有大量的催化活性位。當加入二甲基二硫(DMDS)后,試樣表面無定形結構的焦炭比重明顯增加。而隨著具有芳香結構的大分子硫化物噻吩和苯并噻吩的加入,焦層中基本觀測不到絲狀焦的生成,表明試樣表面的催化活性迅速降低。當裂解時間為3 h,空白試樣表面的焦層呈現絮狀形貌,這主要是由于絲狀焦相互纏繞,對氣相中無定形焦炭產生了“捕捉”作用,這兩類焦炭共同導致了該類形貌的產生[2,4]。當加入二甲基二硫后,隨著裂解時間的延長,焦層展現出粒狀形貌;當芳香結構的硫化物加入后,試樣表面被尺寸3~4 μm的焦粒所覆蓋。
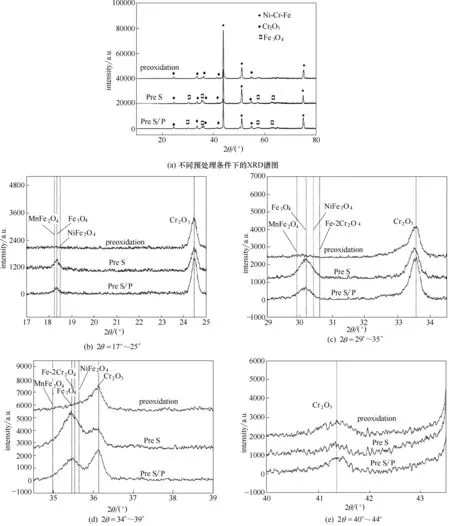
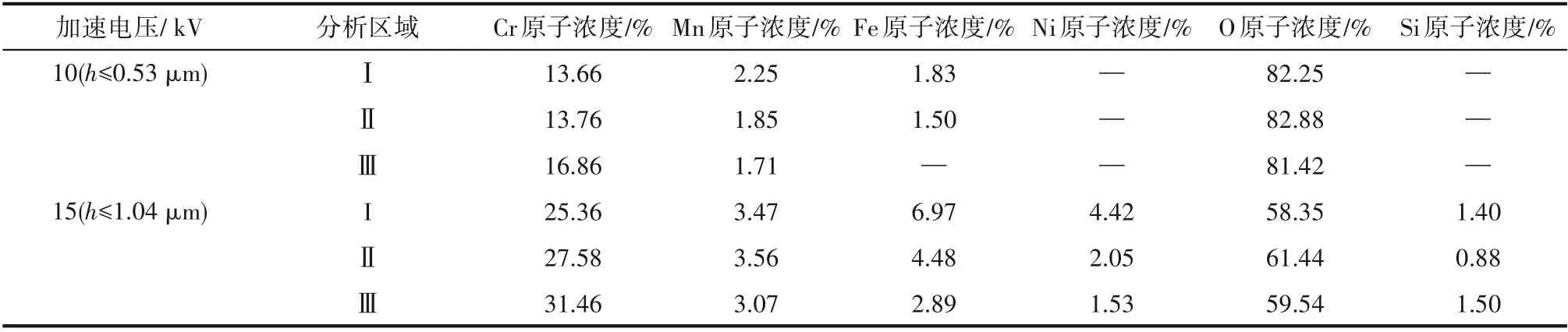
表3 不同預處理條件下HP40合金表面氧化層元素分布Table 3 Element concentrations of the oxide films on HP40 alloys
圖5為添加和未添加二甲基二硫后的試樣表面碳纖維的SEM 圖。由圖5(a)可見,在場發射電鏡下,空白試樣表面的碳纖維具有明亮的端部,表明金屬顆粒被包裹在焦絲中。碳纖維呈現出“分節蠕蟲”狀形貌(segmented worms),這是由于未添加硫化物條件下,碳在催化金屬顆粒邊緣的生成速率大于碳在顆粒中的擴散速率,從而導致節狀纖維的產生[2-3]。由圖5(b)可見,加入硫化物后,“蠕蟲”形貌的纖維碳消失,說明碳纖維生長控制步發生轉變,焦層中的碳纖維呈現螺線狀(spiral filament)交織在一起,這是由于碳在金屬碳化物晶體中擴散路徑長度不同,進而造成金屬顆粒邊緣處的碳纖維生長沿螺線方向進行[2]。與空白試樣相比,硫化物加入條件下,碳纖維直徑明顯增加(約800 nm)。Boehm[16]發現,碳纖維直徑主要由金屬催化顆粒尺寸決定。二甲基二硫在高溫裂解氣氛中的主要產物H2S,會引成基底金屬的晶界脆化,進而造成金屬表層部位抗拉強度降低,加速了尺寸較大的金屬顆粒由表面脫離的過程[2-3]。綜上所述,硫化物對于碳纖維的生長過程具有較大的影響,碳纖維的形貌特征發生了明顯的變化。
實驗方案為sulfides/TEP 和sulfides/TPPI條件下的熱裂解結焦形貌見圖6。其中,二甲基二硫、噻吩與苯并噻吩作為硫化物混合物,質量濃度(以naphtha 質量計)分別為CDMDS=100 μg/g、Cthiophene=89.37 μg/g 和Cbenzothiophene=142.55 μg/g,以保證原料中硫質量分數為Csulfur= 136 μg/g。亞磷酸三乙酯和亞磷酸三苯酯以濃度Ctriethylphosphite=100 μg/g 和Ctriphenylphosphite=100 μg/g 分別與硫化物混合物混合,添加至裂解原料中。由圖6 可見,結焦層主要由無定形結構的熱裂解焦組成,焦炭粒子尺寸約為1~2 μm,表明硫/磷化合物可以有效抑制催化結焦的生長。不同磷化物條件下的焦炭形貌相似,說明兩種混合物的抑制機理相同。與添加硫化物相比(圖4),磷化物加入混合硫化物后,焦炭粒子結合緊密,粒子尺寸減小,焦炭層較為平滑,表明結焦層表面自由基活性位性質發生了改變[5],而亞磷酸三苯酯的添加導致結焦層性質改變更為明顯[圖6(b)]。

圖3 不同預處理條件下HP40合金表面氧化層的SEM照片和EDS分析Fig.3 SEM photos and EDS analysis of the oxide films on HP40 alloys
添加硫化物后,試樣表面焦炭沉積量情況見圖7。可見,在實驗濃度范圍內,不同類型的硫化物均展現了明顯的抑制結焦效果。通常硫化物的添加可以降低爐管催化活性,減少催化結焦的生成,主要是因為高溫下分解形成的HS·自由基可以優先吸附于金屬,從而達到鈍化爐管的效果。當裂解時間為1 h、相同硫質量濃度下,二甲基二硫、噻吩與苯并噻吩的抑焦率達到了64.1%、75.9%和81.4%;當裂解時間為3 h,三者的抑焦率分別為75.7%、82.3%和85.9%。在短期裂解時間內,芳香結構的大分子硫化物具有更為優異的抗結焦性能,這主要是由于芳環硫的大分子幾何特性,使得芳環結構優先與催化活性表面形成化學吸附,進而減少了催化結焦的生成[2,17]。在熱裂解條件下,亞磷酸三乙酯分解生成的(C2H5O)2PO·和(C2H5O)2P·自由基通過與金屬表面形成金屬-磷的復合物(metal-phosphorus complex),從而降低金屬基底活性[5]。亞磷酸三苯酯分解得到的(C6H5O)2PO·和(C6H5O)2P·,在分子級別具有較大的幾何尺寸,因此會與金屬形成尺寸更大的復合物,相比小分子磷化物或衍生物,具有更好的抑制效果。當裂解時間為1 h,方案sulfides/TEP 和sulfides/TPPI的抑焦率達到了82.8%和86.9%;當裂解時間為3 h,兩者的抑焦率分別為87.9%和88.9%。可見,硫/磷化合物由于硫、磷的協同作用,短期內的抑制效果較為顯著。


圖4 原料中添加硫化物后,HP40合金氧化層表面焦炭的SEM圖Fig.4 SEM images of cokes on HP40 alloys with the addition of sulfides(Csulfur=136 μg/g)

圖5 裂解時間1 h條件下,未添加/添加二甲基二硫后HP40合金氧化層表面碳纖維的SEM圖Fig.5 SEM images of carbon fibers on HP40 alloys in the absence/presence of DMDS after cracking time of 1 h

圖6 裂解時間3 h條件下,添加硫化物/磷化物混合物后HP40合金氧化層表面焦炭的SEM圖Fig.6 SEM images of cokes on HP40 alloys in the presence of S/P compounds after cracking time of 3 h
2.3 試樣表面預處理對熱裂解結焦行為的影響
實驗方案為Pre S/P 條件下,試樣表面的焦炭形貌見圖8,其中,預處理工藝見表1。當裂解時間為1 h,氧化試樣表面仍然發現了絲狀焦炭,絲狀焦由基底向外伸出[圖8(c)],說明硫/磷預處理方式不能完全抑制催化結焦的生成。對絲狀焦炭進行EDS分析可知,Fe為主要催化顆粒[圖8(d)]。當裂解時間為3 h,結焦層主要由無定形結構焦炭組成,表明此時試樣表面催化活性下降,金屬催化粒子或被結焦層覆蓋。
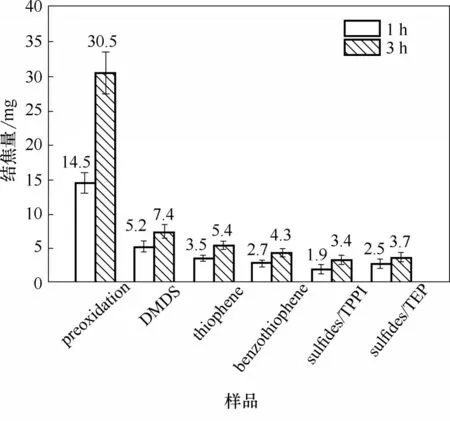
圖7 裂解時間1 h和3 h條件下,添加硫化物后HP40合金表面焦炭沉積量情況Fig.7 The amounts of coke formed on HP40 alloys with the addition of sulfides after cracking time of 1 h and 3 h
實驗方案為Pre S條件下,試樣表面的焦炭形貌見圖9。當裂解時間為1 h,焦層中的絲狀焦炭在Fe顆粒的催化作用下由基底向外生長[圖8(d)]。當裂解時間為3 h,試樣表面布滿了無定形結構焦炭。與圖8(c)相比,圖9(c)中絲狀焦直徑增加。在相同裂解條件下,碳纖維直徑主要由金屬催化顆粒尺寸決定[16]。絲狀焦直徑的變化說明,磷化物的加入在金屬表面形成了較為穩定的鈍化膜,降低了硫化物對于基底金屬晶界脆化的影響,延緩了大尺寸金屬顆粒由試樣表面脫離的過程。
圖10 為不同預處理下試樣表面焦炭沉積量情況。當裂解時間為1 h 時,Pre S/P 和Pre S 兩種預處理方法保持了一定的抑制結焦效果,抑制率分別為40%和29.7%;裂解時間為3 h,抑制率分別為39.7%和26.2%。與原料中連續注入硫化物或硫/磷化合物的方式相比,預處理抑制結焦效果有限,主要原因為預處理過程中產生的HS·在金屬表面的吸附為動態過程,在后續的高溫裂解氣氛中,吸附態硫易于脫附,從而導致活性金屬表面重新暴露于裂解氣氛中,合金試樣表面的鈍化效果隨裂解時間的延長而下降。由試樣表面表征結果可知,預處理未導致氧化表面顯微結構發生明顯變化,但表面氧化層中Fe 含量升高,并在隨后裂解過程中成為絲狀焦生長的主要催化活性元素[圖8(d)和圖9(d)]。預處理中磷化物的加入,可以減少較大的金屬顆粒從氧化表面的脫離,并降低初始階段結焦速率,但為了維持較高的抑制效果,仍需要采用原料連續注入的方法。
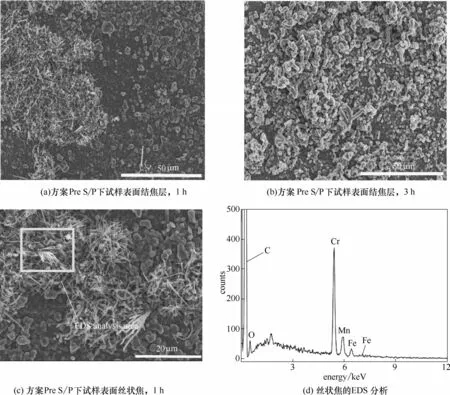
圖8 裂解時間1 h和3 h、實驗方案為“Pre S/P”條件下,HP40合金表面焦炭的SEM圖和絲狀焦的EDS分析Fig.8 SEM images of cokes on HP40 alloys under the condition of“Pre S/P”after cracking time of 1h and 3 h;EDS of cokes on HP40 alloys

圖9 裂解時間1 h和3 h、實驗方案為Pre S條件下,HP40合金表面焦炭的SEM圖和表面絲狀焦的EDS分析Fig.9 SEM images of cokes on HP40 alloys under the condition of Pre S after cracking time of 1h and 3 h;EDS of cokes on HP40 alloys
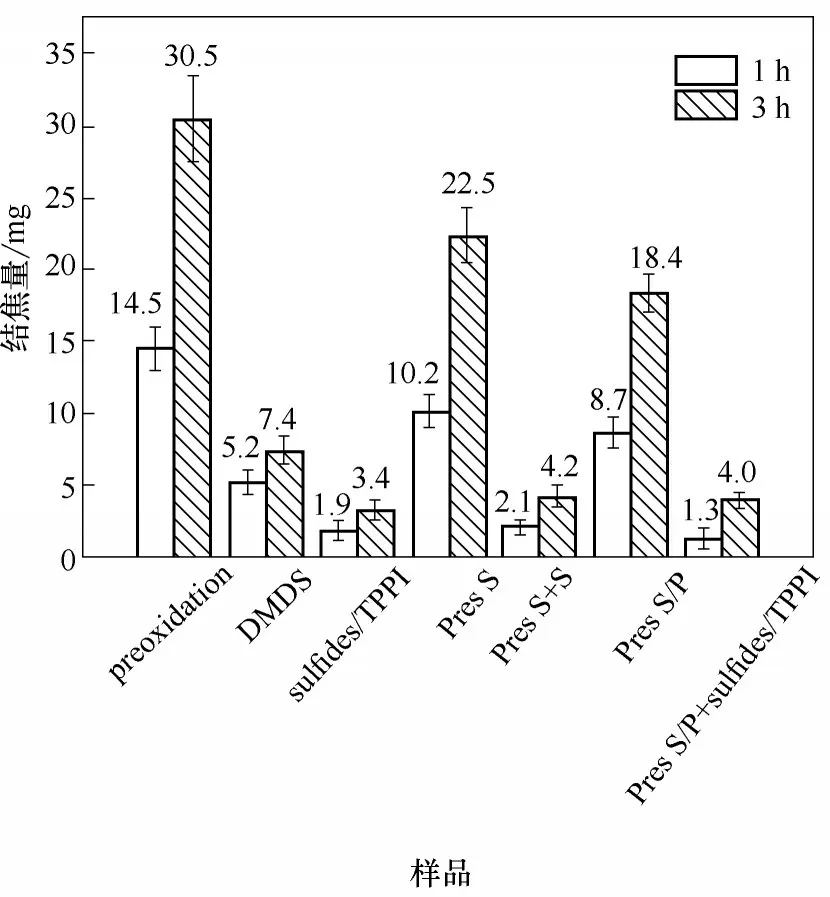
圖10 裂解時間1 h和3 h條件下,不同添加方式對HP40合金表面焦炭沉積量的影響Fig.10 The amounts of coke formed on HP40 alloys with varying the addition methods after cracking time of 1 h and 3 h
2.4 表面預處理與原料連續注入聯合方式對熱裂解結焦行為的影響

圖11 裂解時間1 h和3 h、實驗方案為Pre S+S條件下HP40合金表面焦炭的SEM圖和表面絲狀焦的EDS分析Fig.11 SEM images of cokes on HP40 alloys under the condition of Pre S+S after cracking time of 1 h and 3 h;EDS of coke on HP40 alloys
圖11 為方案Pre S + S 操作方式下試樣表面的焦炭形貌,其中硫化物預處理條件見表1,DMDS 的連續添加濃度為200 μg/g(以naphtha 質量計)。當裂解時間為1 h,試樣表面布滿了無定形結構焦炭,焦炭粒子結合疏松,焦層中僅發現了少量的尺寸細小的針狀焦。與硫化物預處理[圖9(a)]和原料連續注硫[圖4(c)]兩類方式相比可知,聯合操作方式有效地降低了金屬表面催化活性,抑制催化結焦的形核與生長。當裂解時間為3 h,試樣表面的無定形結構焦炭顆粒結合緊密,但結焦層中出現了棒狀焦,并由焦層內部向外生長。雖然原料連續注硫可以抑制催化結焦的生成[圖4(d)],但前期的預硫化處理過程會導致氧化層表面FexOy含量的增加,且吸附態硫易于脫附。最終,隨著裂解過程的進行,催化結焦重新在活性表面成核并生長。EDS 分析可知,棒狀焦炭中金屬元素的信號較弱,這可能是金屬顆粒還未從金屬基底脫離[3]。
圖12為Pre S/P+sulfides/TPPI 操作方式下試樣表面的焦炭形貌。硫/磷組分預處理條件見表1,裂解原料中各添加組分質量濃度與實驗方案sulfides/TPPI 相同。當裂解時間為1 h,結焦層由無定形結構的焦炭粒子組成,未發現絲狀焦。當裂解時間為3 h,焦炭粒子結合緊密,僅有少量的催化焦絲向外伸出。絲狀焦的出現主要是因為硫/磷化預處理導致氧化層表面FexOy含量的增加,隨著裂解時間的延長,催化結焦開始在催化活性表面生長。由焦層形貌對比發現[圖10(b)和圖11(b)],與Pre S + S 方式相比,磷的加入更有效地抑制催化結焦的生長,該條件下的結焦層較為平滑,無定形結構焦炭粒子尺寸和催化焦直徑均有減小。
不同聯合處理條件下,試樣表面焦炭沉積量情況見圖10。當裂解時間為1 h,方案Pre S+S 和Pre S/P+sulfides/TPPI 的抑制率分別為85.5%和91.0%;當裂解時間為3 h,抑制率分別為86.2%和86.9%。在預處理過程中,HS·自由基通過化學吸附使金屬活性位中毒,磷化物自由基通過聚合反應可以形成熱穩定性優異的聚磷酸鹽層(polyphosphate layer),并吸附于金屬表面[18],在隨后的裂解過程,硫/磷化合物的連續加入為該過程進一步提供源物質補充。因此,在所有添加方式中,方案Pre S/P + sulfides/TPPI 會使金屬表面催化活性位被最大程度地覆蓋,在裂解初期顯示出最為優異的抗結焦效果。當裂解時間為3 h,該方案下的結焦量略高于方案sulfides/TPPI,這可能是因為表面活性位被覆蓋后,連續加入的硫/磷化合物自由基會吸附于碳基體,從而提高結焦層表面的自由基活性位的數目,促進結焦母體與焦層表面之間的非異相自由基反應(heterogeneous free-radical reaction),使結焦量增加[7,19]。在整個實驗周期,兩種聯合處理的抗結焦效果相近,但方案Pre S+S下的催化結焦含量更高,而且無定形焦炭粒子的直徑較大,說明磷化物的加入不僅提高了預處理的效果,而且改變了結焦層的結構性質。

圖12 裂解時間1 h和3 h條件下,硫/磷組分預處理與原料連續注入硫/磷化合物聯合操作方式下HP40合金表面焦炭的SEM圖Fig.12 SEM photos of cokes on HP40 alloys with the pretreatment of DMDS/TPPI followed by continuous addition of mixed sulfides/TPPI after cracking time of 1 h and 3 h
2.5 熱裂解結焦的結構分析
圖13為裂解時間為3 h下焦炭的拉曼光譜。可見,不同添加方式下所形成的熱裂解焦炭的Raman光譜均展示了相似的峰形,包括了非晶碳材料的一階和二階光譜。其中,1350 cm-1和1580 cm-1附近處出現的兩個較強的寬峰為碳材料的一階光譜[圖13(a)],2500~3300 cm-1的寬峰對應于二階光譜[圖13(b)][20-21]。利用Origin9.0軟件對熱裂解結焦的拉曼光譜進行分峰擬合,擬考察添加方式對熱裂解結焦結構的影響,擬合參數見表4,擬合圖譜結果見圖13(c)和圖13(d)。
由圖可知,熱裂解結焦的一階拉曼光譜區域由五個峰疊加而成。G 峰是石墨狀sp2雜化C—C鍵散射的結果,來源于sp2鍵的所有伸縮振動模式(芳香環和烯烴結構)。D 峰群(D1~D4 峰)是無序碳所共有的特征,源于芳香環式結構的伸縮振動模式。二階拉曼光譜區域的寬峰可能來自于一階光譜峰的泛音及振動模式的組合[20]。分峰擬合結果見表5,其中,峰面積強度比ID1/IG可作為材料石墨化程度的評判標準,ID3/IG可用來定性評價結焦層中無定形結構焦炭含量[7]。由表可知,在所有添加方式下,G 峰寬度不僅比D1 峰窄,峰面積也較小,表明組成熱裂解焦的碳原子排列的有序程度較差。D3與D4峰寬度比G 峰寬,表明焦層中存在大量的無定形結構焦炭[22],這也與SEM形貌結果吻合。在所有樣品中,空白樣品的G峰面積相對值IG/IAll最大,而D1峰面積與G 峰面積比值ID1/IG最小,表明該條件下焦炭石墨化程度最高[9]。ID3/IG比值結果表明,不同添加方式都會引起無定形焦炭含量的增加,這與SEM 形貌結果一致。在方案Pre S/P + sulfides/TPPI 條件下,焦炭的ID1/IG和ID3/IG值最大,說明硫/磷化合物自由基不僅通過鈍化金屬降低了有序化程度較高的催化結焦數量,在后續連續添加過程中,自由基進一步改變了碳層表面活性位數目、增加了石墨晶格間隙(interstitial space)[7,9],從而導致碳原子排列的無序化程度增加,無定形結構焦炭的含量增加,石墨化程度降低。由表4 可知,原料連續添加噻吩與苯并噻吩條件下,熱裂解焦炭的ID1/IG比值大于連續添加二甲基二硫下的比值,說明了噻吩類硫使碳的結構缺陷增加。硫摻雜碳材料制備方面的研究發現[23-24],高溫條件下(>700℃),噻吩類硫可以穩定地摻雜進入sp2雜化的碳晶格,導致碳基體結構缺陷增加。活性碳在燃料油深度脫硫應用方面的研究還發現[25-26],噻吩類硫化物芳環中的π 電子會與石墨表面富電子區域優先形成π-π 復合結構(π-π complex),從而導致硫化物分子在石墨平面的優先吸附,進而對石墨平面結構(graphene sheets)的構建過程造成影響。而芳環結構在進一步縮合脫氫過程中,硫極有可能繼續以噻吩環的形式存在于碳晶格邊緣或缺陷部位,改變結焦層表面活性位特性[27]。因此,硫化物特性的差異會造成硫在熱裂解焦層中分布及存在形式上的差別,從而導致結焦層化學結構發生變化。

表4 熱裂解結焦一階拉曼光譜的分峰擬合參數[20-21]Table 4 The parameters of curve fits for the first-order Raman spectra of cokes on HP40 alloys
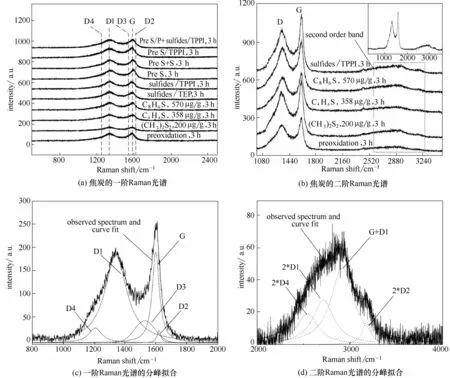
圖13 不同添加方式下HP40合金表面焦炭的Raman光譜圖Fig.13 Raman spectra of cokes on HP40 alloys with varied addition methods
圖14 為連續添加DMDS 及硫/磷化合物條件下試樣表面焦炭的C 1s XPS 譜圖。可見,焦層中碳的C 1s 峰位不對稱(以284.5 eV 為中心),這是石墨碳sp2雜化的結果。采用XPSPEAK 軟件對碳譜分峰擬合,主要峰位包括C C sp2(284.5 eV)、C—C sp3(285.2 eV)、C—O(286.8 eV)和—HO—C O(288.1 eV)[28]。由圖可知,硫化物及硫/磷化合物連續添加條件下,譜圖中C—C 峰強度上升,意味結焦層中無定形碳含量升高[28]。方案sulfides/TPPI下,結焦層中C—O含量增加,這主要是因為所添加的磷化物分子結構中含氧。XPS 譜圖中,Gaussian 曲線的峰面積反映了它所對的形態在樣品中的相對含量。表6為硫化物及硫/磷化合物加入條件下焦炭的sp3/sp2比值。可見,空白樣品的比值最小,表明焦層中sp3含量最小,其結構的有序化程度最高[28]。
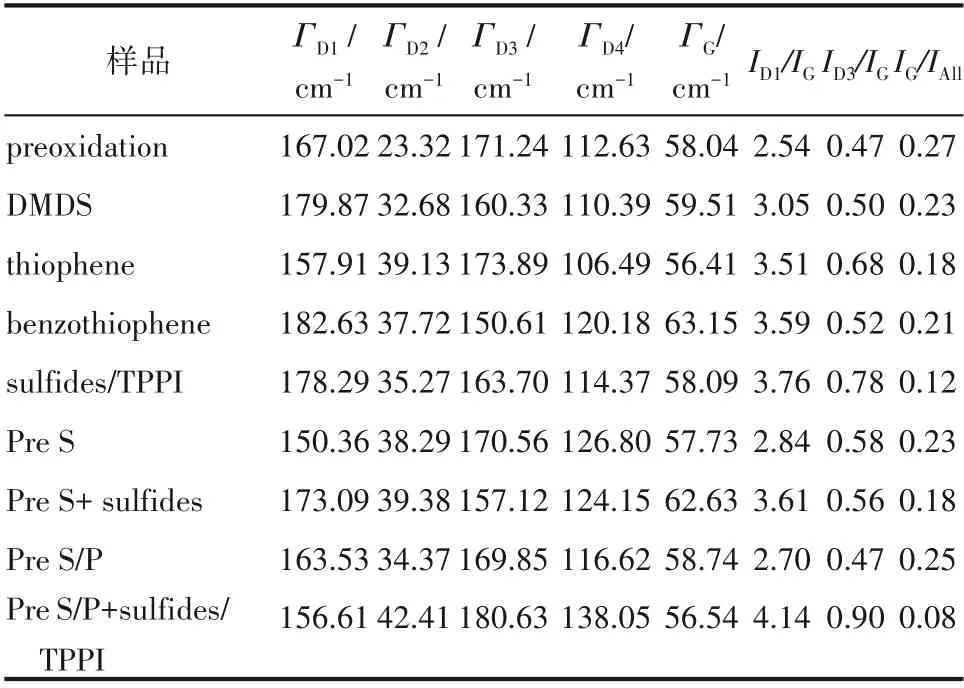
表5 熱裂解結焦拉曼光譜的分峰擬合結果Table 5 Fitting results of Raman spectra of coke deposits
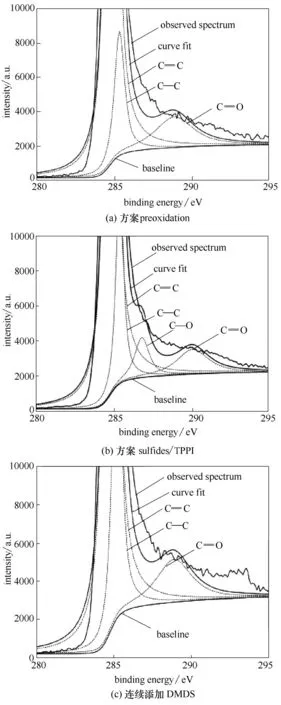
圖14 連續添加DMDS及硫/磷化合物條件下HP40合金表面焦炭的C 1s XPS光譜圖Fig.14 C1s XPS spectra of cokes on HP40 alloys with the continuous addition of DMDS and sulfides/TPPI

表6 熱裂解結焦的sp3/sp2比值Table 6 sp3/sp2 ratio of coke deposits
裂解時間為3 h,不同添加方式下焦炭的氫/碳比情況見表7。測試結果表明:不同添加方式下焦層中C 元素含量均在95%以上,H 元素含量在0.7%~3%。烴類裂解過程中,結焦母體在聚合生焦過程不斷脫氫,導致焦炭氫含量降低,焦炭的縮合度增加。其中,Fe、Ni 等金屬粒子會進一步催化碳纖維脫氫,因此,由金屬催化形成的絲狀焦炭氫含量極低[2]。由表7可知,空白試樣表面焦炭的氫含量最低,表明催化結焦現象嚴重。焦炭的碳氫原子比大于8.0,說明熱裂解焦縮合度較高[32]。而原料加入硫化物和硫/磷化合物后,焦炭的氫/碳比升高,表明化合物通過鈍化金屬表面催化活性位,在一定程度上減少了催化結焦的生成。

圖15 連續添加硫/磷化合物條件下HP40合金表面焦炭的S 2p和P 2p XPS光譜圖Fig.15 S 2p and P 2p XPS spectra of cokes on HP40 alloys with the continuous addition of sulfides/TPPI

表7 熱裂解結焦的H/C比值(質量比)Table 7 The H/C mass ratios of coke deposits
3 結 論
(1)經過空氣氧化、硫化物和硫/磷化合物預處理后,HP40合金表面主要生成了Cr2O3和FexOy等氧化物結構,三種處理條件下的氧化物成分類似。經過硫化物或硫/磷化合物預處理后,試樣表面氧化物主要呈現片狀和粒狀形貌。與空氣氧化方式相比,硫化物和硫磷化合物預處理導致氧化層中Fe含量升高。
(2)在原料連續添加硫化物條件下,試樣表面無定形結構的焦炭比重增加,芳環硫加入時,芳環分子結構優先與催化活性表面形成化學吸附,明顯減少了催化結焦的生成;磷化物加入混合硫化物后,由于硫/磷化合物的協同作用,顯示出更為優異的抗結焦效果,磷化物自由基改變了結焦層表面自由基活性位性質,使得焦炭粒子尺寸減小,焦炭層較為平滑。
(3)預硫化和硫/磷預處理的抑制結焦效果有限,主要是因為預處理過程中HS·在金屬表面易于脫附,從而導致試樣的鈍化效果隨裂解時間下降,而且預處理造成氧化層中Fe含量升高,并在隨后裂解過程中進一步催化絲狀焦的生長。磷化物的加入可以在金屬表面形成穩定的鈍化膜,阻礙大尺寸金屬顆粒由試樣表面脫離。
(4)在裂解初期,預硫化與原料連續加硫的聯合操作方式有效地降低了金屬表面催化活性,但前期預硫化導致氧化層表面Fe含量增加,催化結焦隨著裂解過程的進行重新開始生長。硫/磷預處理與原料連續注入硫/磷化合物聯合操作方式與原料連續注入硫/磷化合物的抗結焦效果接近,但前者在初期的抑制效果更明顯。兩種聯合處理的抗結焦效果相近,磷化物的加入不僅提高了預處理的效果,而且改變了結焦層的結構。
(5)熱裂解結焦的Raman 光譜結果顯示,所有操作方式都會引起結焦層中無定形焦炭含量升高,焦炭的石墨化程度降低。“硫/磷組分預處理+連續注入硫/磷化合物”聯合條件下,焦炭的ID1/IG和ID3/IG值最大,說明該條件下碳原子排列的無序化程度最高,無定形焦炭含量較高。熱裂解焦炭縮合程度較高,硫化物和硫/磷化合物減少了催化結焦的生成,在一定程度上提高了焦層中氫含量。
