高效液相色譜-串聯質譜法測定飼料中喹乙醇的不確定度評估
2020-12-16 13:18:30楊彥寧歐海軍譚美英隆雪明周燦陳福華
飼料博覽 2020年9期
楊彥寧,歐海軍,譚美英,隆雪明,周燦,陳福華
(湖南省獸藥飼料監察所,長沙410000)
隨著“造肉1號“味霸”“速肥肽”等非法飼料添加劑的曝光,“喹乙醇”逐步進入公眾視野。這種被稱為“新型瘦肉精”的藥物,是1965年由德國人合成的一種抗菌助生長劑[1],具有蛋白同化作用,可提高飼料轉化率與瘦肉率,促進動物生長。但大劑量的使用喹乙醇會對動物產生毒性作用[2]。喹乙醇在體內具有一定蓄積性,可對動物或人產生致畸、致癌、致突變[3]。歐洲和美國已經禁止喹乙醇作為飼料添加劑使用。我國農業農村部第2638號公告也明確表示,由于喹乙醇獸藥的原料藥及各種制劑可能對動物產品質量安全、公共衛生安全和生態安全存在風險隱患,根據《獸藥管理條例》第六十九條規定,決定停止在食品動物中使用喹乙醇。因此,喹乙醇檢測結果的準確性在保障動物源性食品安全和公共衛生安全,確保養殖環節依法、科學、合理用藥,為獸藥執法監管工作的有效開展提供有力的支撐時顯得尤為重要。
本文采用高效液相色譜-串聯質譜測定飼料中喹乙醇,采用JJF-1059-2012《測量不確定度評定與表示》、JJF1135-2005《化學分析測量不確定評定》、CNAS-GL006-2019《化學分析中不確定度評估指南》等對結果進行不確定度評估,對不確定度各分量進行計算,探究影響檢測結果不確定度的影響因素,為評定檢測結果的準確性提供參考[4-9]。
1 材料與方法
1.1 儀器與試劑
高效液相色譜-串聯質譜(AB 3200Q Trap):配有電噴霧離子源(ESI),固相萃取裝置,喹乙醇標準品(Dr),HLB固相萃取小柱(200 mg/6 mL),甲醇、乙腈、甲酸(色譜純)。
1.2 檢測方法
依據農業部2086號公告-5-2014《飼料中卡巴氧、乙酰甲喹、喹烯酮和喹乙醇的測定液相色譜-串聯質譜法》進行檢測。
1.3 樣品制備
準確稱量預混合飼料1 g(精確至0.01 g),加入0.1%甲酸-乙腈溶液10 mL,渦旋1 min,40℃超聲波清洗儀中超聲10 min,9 000 r·min-1離心15 min,收集上清液。殘渣用0.1%甲酸-乙腈溶液10 mL重復提取1次,合并提取液,混勻,準確量取5.0 mL總提取液于10 mL試管中,60℃氮氣吹至約2 mL,加入0.1 mol·L-1磷酸二氫鉀溶液,充分溶解殘余物,為備用液。轉移至已活化的HLB固相萃取柱,待試樣完全通過固相萃取柱后用0.02 mol·L-1鹽酸3 mL和5%甲醇3 mL淋洗,擠干,用5 mL甲醇洗脫,收集洗脫液并氮氣吹干,用20%乙腈溶液1 mL充分溶解,過0.22μm濾膜后上高效液相色譜-串聯質譜儀(LC-MS/MS)檢測。
1.4 儀器條件
1.4.1色譜條件
色譜柱ACQUITY UPLC BEH C18柱(2.1 mm×100 mm 1.7μm),柱溫為40℃;進樣體積10μL流速為0.5 mL·min-1,流動相A乙腈,流動相B為0.1%甲酸水溶液,梯度為0~0.5 min,體積分數為5%的B溶液;0.5~4 min,體積分數為5%~90%的B溶液,4~6 min,體積分數為90%的B溶液,6~7 min,體積分數為90%~5%的B溶液。
1.4.2質譜條件
離子化模式為電噴霧正離子模式,質譜掃描方式為MRM,離子源溫度為600℃、碰撞能量及定性定量離子質譜參數如表1所示。

表1喹乙醇質譜參數及保留時間
2 結果與討論
2.1 數學模型
采用外標法定量,按下式計算喹乙醇含量:

式中,X為樣品中待測組分含量(mg·kg-1);C被測樣品濃度(μg·mL-1);V1提取液定容體積;V2用于SPE凈化的提取液體積,5 mL;V3最終上機的濃縮液體積,1 mL;m樣品質量(g)。上述公式依據樣品測定原理給出,未考慮隨機因素對測量結果的影響。
2.2 不確定度來源
根據檢測過程和喹乙醇含量的計算公式,得到不確定度來源圖,見圖1。
2.3 各分量標準不確定度的評估
2.3.1測量重復性的相對標準不確定度
由于分析過程中,很多操作引入的不確定度具有一定的重復性,如天平稱量,移液管轉移液體,容量瓶定容體積,回收率等操作等,因此,可以將此類分量進行歸納,引入重復性,即精密度,用于分析研究過程中隨機變化引入的不確定度。精密度主要采用統計學方式進行研究,對加標樣品中喹乙醇含量進行一系列平行測試,得到分析過程總隨機變化,以該標準化差值的標準偏差評估試驗過程的重復性不確定度。
在重復性條件下,對同一加標樣品中喹乙醇的含量進行獨立8次測量,結果見表2,綜合上述數據,按照貝塞爾公式計算測量結果的標準差,單次測量的標準不確定度為:
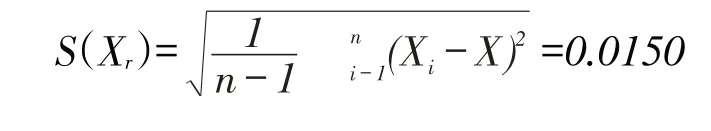
其算數平均值的標準不確定度為:

重復條件下,喹乙醇含量的相對標準不確定度為:

上述分量合并研究各種隨機因素,如體積讀數偏差、LC-MS/MS變動,樣品混合不均勻以及HLB固相萃取小柱對回收率的影響等,后續評定其他分量時,不再考慮各種隨機因素。
2.3.2試樣稱量引入的不確定度
試樣稱量引入的不確定度主要是天平的校準,允許誤差±0.001 g,假定為均勻分布,其標準不確定度為:

預混合飼料1 g,則其相對標準不確定度為:
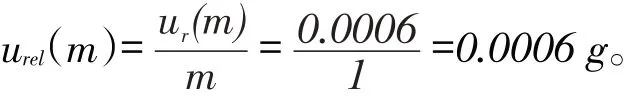
2.3.3提取液V1引入的不確定度
用10 mL單標吸管吸取10 mL 0.1%甲酸-乙腈提取液,根據檢定規程JJG196-2006[8],10 mL A級吸管允許誤差為±0.02 mL,假定為三角分布,則10 mL的單標吸管的相對標準不確定度為:

由于方法中,需重復提取1次,兩次移取獨立不相關,因此,提取液引入的不確定度合成得:

圖1喹乙醇檢測過程中不確定度來源

表2加標飼料中喹乙醇含量mg kg-1
2.3.4凈化液V2引入的不確定度
從提取液中量取5.0 mL待氮吹、凈化,根據檢定規程JJG196-2006,5 mL A級單標吸管允許誤差為±0.015 mL,假定為三角分布,則5 mL單標吸管的相對標準不確定度為:
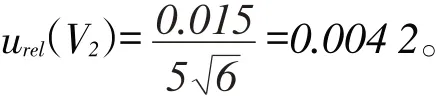
2.3.5用于測定樣品定容體積V3引入的不確定度
用1 mL的單標吸管量取20%乙腈溶液充分溶解已吹干試樣,根據檢定規程JJG196-2006,1 mL A級單標吸管的允許誤差為±0.007 mL,假定為三角分布,1 mL的單標吸管的相對標準不確定度為:
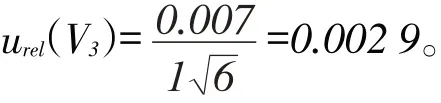
2.3.6標準品引入的不確定度
標準品純度引入的不確定度:方法中采用的喹乙醇標準品,純度為97.89%,根據證書提供的不確定度為0.73%,假定均勻分布,則其不確定度為:

標準品稱量引入的不確定度:準確稱量標準品喹乙醇8.523 85 g,稱量時使用分辨率0.000 01 g分析天平,由2.3.2可知,其相對標準不確定度為:

標準儲備液定容引入的不確定度:標準品用甲醇定容至10 mL,使用容量瓶為10 mL A級,根據檢定規程JJG196-2006,10 mL A級容量瓶的允許誤差為±0.02 mL,假定為三角分布,則10 mL的容量瓶的標準不確定度為:

綜上,在標準液配制時引入的不確定度由上述三種合成,即

使用標準溶液因溫度變動引起的標準溶液濃度不確定度:室溫(20±5℃),甲醇的膨脹系數為1.1×10-3/℃,則由溫度引起的體積變化為±5×1.1×10-3=±0.005 05 mL,假定均勻分布,則:u(T)=標準品引入的不確定度由以上不確定度合成,由標準溶液引入的不確定度為:

2.3.7測定液引入的不確定度
本次人工配制濃度為0.834 4 mg·mL-1喹乙醇儲備液,稀釋為62.58、104.3、208.6、417.2、834.4 ng·mL-1標準系列,引入不確定度,校正曲線為Ai=a+bCi,由工作曲線變動性引起濃度c的標準不確定度分量為:校正曲線各個參數如表3所示。

表3校正曲線參數
2.4 合成標準不確定度
2.4.1相對不確定度
各分量的相對標準不確定度見表4。

表4相對不確定度各分量
按照評定不確定度的測量模型計算喹乙醇含量的合成不確定度,

本次測定,樣品中喹乙醇為0.763 mg·kg-1,則
2.4.2擴展不確定度
根據國際慣例95%置信概率取包含因子K=2,則擴展不確定度為u(x)=k×u=2×0.012 2=0.024 4 mg·kg-1。
2.5 檢測結果的表示
用液相色譜-串聯質譜測定飼料中喹乙醇,結果表示為(0.763±0.0244)mg·kg-1,其中擴展系數為k=2。
3 結果分析
由相對不確定度直方圖(見圖2)可以看出,對喹乙醇結果影響最大的是urel(V1),即提取液的體積,其次是重復性操作urel(fr)及標準溶液的配制urel(s),因此,在對飼料中喹乙醇含量進行檢測時,應盡量確保提取液體積移取準確,標準溶液配制及稀釋精準,在條件允許情況下,多作平行樣,保證重復性操作的規范性,消除系統誤差影響。
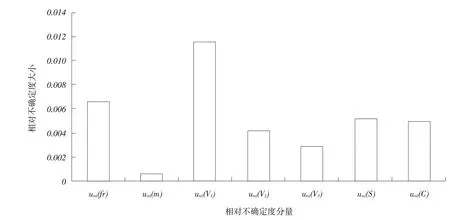
圖2相對不確定度直方圖
4 結論
通過分析高效液相色譜-串聯質譜測定飼料中喹乙醇不確定度來源,對各不確定度分量進行計算,得出當飼料中喹乙醇含量為0.763 mg·kg-1時,其擴展不確定度為0.0244 mg·kg-1(k=2)。通過對其中不確定分量進行分析發現,影響檢測結果的主要因素是提取液體積、重復性操作、以及標準品的配制與稀釋,因此,在實際檢測中,應嚴格控制試驗條件,保證移液管等規范使用,標準溶液配制準確無誤,也應加強檢驗人員崗位培訓,確保試驗過程規范性和重現性,減小此類不確定度分量對檢測結果帶來的影響,也為飼料質量安全控制提供有效、可靠、可溯源的測量數據。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
當代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
海峽科技與產業(2016年3期)2016-05-17 04:32:12
