固相萃取-火焰原子吸收光譜法分析水溶肥料鉻形態
2021-01-18 04:21:16韓巖松黃均明保萬魁劉紅芳
中國土壤與肥料 2020年6期
關鍵詞:實驗
韓巖松,黃均明,保萬魁,劉紅芳,劉 蜜,王 旭
鉻是第四周期ⅥB族元素,在地殼中平均含量0.010%~0.011%。自然界中的鉻廣泛存在于巖石、土壤、大氣、水及生物體中,可形成從+2到+6多種價態化合物,最常見的是Cr3+和Cr6+化合物。近代醫學研究表明,不同價態的鉻會產生不同的生理作用。Cr3+是人體維持生命的必需元素,對于維持葡萄糖[1-2]、脂質和蛋白質的代謝具有重要作用;而Cr6+在生物體內有很強的流動性,以CrO42+和Cr2O72-的形態促使細胞膜氧化,從而導致病變發生,其毒性一般為Cr3+毒性的100~ 1 000倍[3]。鉻的形態分布影響著鉻在環境中的遷移轉化規律。因此,鉻的形態分析對于環境質量評價、生態效應和致毒機理研究具有重要意義。
近年來,國內外關于水質鉻形態分析的分離和檢測手段已有不少報道,其中分離富集方法有濁點萃取(CPE)[4-6]、離子交換法[7-8]、高效液相色譜法(HPLC)[9]、毛細管電泳(CE)[10]等。由于Cr6+和Cr3+在水溶液中荷電性的不同,使得離子交換法分離鉻形態時具有較高的選擇性,同時離子交換分離法還具備富集倍數高、操作簡單等優點。在鉻形態分析的檢測方法中有電化學 法[11-13]、分光光度法[14-21]、原子光譜法[22-23]、中子活化分析法[24]、色譜法[25]、熒光法[26]等。其中使用最多的是原子吸收光譜法(AAS)。與電熱原子吸收光譜法(ETAAS)相比,火焰原子吸收光譜法(FAAS)的靈敏度不是很高,但其具有儀器便宜、操作簡便、重現性好等優點,因此得到了廣泛的應用。
本實驗研究了陰離子交換樹脂柱對水溶肥料中水溶態Cr3+和Cr6+的分離效果,通過優化實驗條件,實現了Cr3+和Cr6+的相互分離,用FAAS法測定Cr3+含量及總鉻含量,Cr6+含量則通過差減法獲得。此方法適用于僅含Cr3+和Cr6+的水溶肥料中鉻形態分析。
1 材料與方法
1.1 試劑
1 mg/mL Cr3+標準儲備液[GSB 04-1723-2004(e)]、1 mg/mL Cr6+標 準 儲 備 液[GSB 04-1723-2004(b)]:國家有色金屬及電子材料分析測試中心,其中Cr3+儲備液為酸性介質,Cr6+儲備液為水介質。
焦硫酸鉀溶液(100 g/L);甲醇,優級純;PAX 60 mg,3 mL陰離子交換柱。
1.2 儀器
ICE 3000 Series原子吸收分光光度計:配備鉻空心陰極燈,燈電流6 mA,測定波長357.9 nm;乙炔燃氣流量1.4 L/min;空氣助氣流量5.5 L/min;光譜通帶0.5 nm;燃燒器高度8.0 mm;恒溫振 蕩器。
1.3 供試樣品
大量元素水溶肥料、微量元素水溶肥料、含氨基酸水溶肥料、含腐植酸水溶肥料、有機水溶肥料等水溶肥料。供試樣品信息見表1。

表1 供試樣品信息
1.4 樣品前處理
1.4.1 試樣的制備
將粉劑樣品縮分至約100 g,將其迅速研磨至全部通過0.50 mm孔徑試驗篩(如樣品潮濕,可通過1.00 mm孔徑試驗篩),混合均勻,置于潔凈、干燥容器中;液體樣品經搖動均勻后,迅速取出約100 mL,置于潔凈、干燥容器中。
1.4.2 試樣溶液的制備
1.4.2.1 水溶態總鉻溶液的制備 固體試樣:稱取1~5 g試樣(精確至0.000 1 g)置于50 mL容量瓶中,加約40 mL水,置于(25±5)℃振蕩器內,在(180±20)r/min頻率下振蕩30 min。取出,加入焦硫酸鉀溶液5 mL,用水定容并搖勻,干過濾,棄去最初幾毫升濾液后,濾液待測。
液體試樣:稱取1~5 g試樣(精確至0.000 1 g)置于50 mL容量瓶中,加入焦硫酸鉀溶液5 mL,用水定容并搖勻,干過濾,棄去最初幾毫升濾液后,濾液待測。
1.4.2.2 水溶態Cr3+溶液的制備 取適量1.4.2.1中的過濾液,緩慢通過陰離子交換樹脂柱,收集濾液,待測。
離子交換樹脂柱預處理采用3 mL甲醇、3 mL水以1.0 mL/min流速各沖洗一次。
1.5 標準曲線的繪制
分別吸取1 mg/mL Cr6+標準溶液0.00、1.00、2.00、4.00、6.00、8.00、10.00 mL于7個100 mL容量瓶中,加入10 mL焦硫酸鉀溶液,用水定容,混勻。該標準系列溶液質量濃度分別為0、0.50、1.00、2.00、3.00、4.00、5.00 μg/mL。在選定最佳工作條件下,于波長357.9 nm處,使用富燃性空氣-乙炔火焰,以鉻含量為0的標準溶液為參比溶液調零,測定各標準溶液的吸光值。
以各標準溶液鉻的質量濃度(μg/mL)為橫坐標,相應的吸光值為縱坐標,繪制曲線。
1.6 試樣溶液的測定
將試樣溶液或經稀釋一定倍數后在與測定標準系列溶液相同的條件下測定,在標準曲線上查出相應的質量濃度(μg/mL)。
測得Cr3+含量及總鉻含量后,Cr6+含量通過差減法獲得。
2 結果與分析
2.1 儀器條件選擇
空氣-乙炔火焰原子吸收法測定鉻,受火焰狀態和燃燒器高度的影響很大。由于鉻的化合物在火焰中易生成難于熔融和原子化的氧化物,因此,選用具有較強還原性能的富燃性火焰進行測定。實驗在測定波長為357.9 nm,鉻空心陰極燈電流為6 mA,光譜通帶為0.5 nm的條件下,研究了燃燒器高度和乙炔/空氣燃助比對鉻測定的影響。
2.1.1 燃助比測定
固定空氣(助氣)的流量為5.5 L/min,通過改變燃氣(乙炔)流量來繪制燃助比曲線(圖1)以確定最佳燃助比。結果表明最佳燃助比為1.4/5.5,火焰狀態為富燃性火焰。
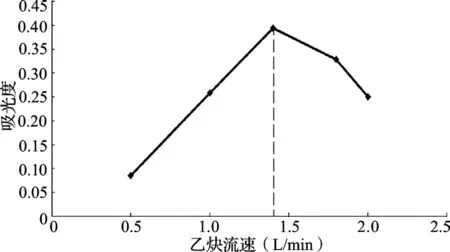
圖1 燃助比曲線
2.1.2 燃燒器高度的確定
實驗研究了不同燃燒器高度對鉻吸光度測定的影響(圖2),結果表明燃燒器高度在7.0~ 9.0 mm范圍內的吸光度較大。實驗選用燃燒器高度為8.0 mm。

圖2 燃燒器高度曲線
2.2 標準曲線的選擇
由于Cr3+標準溶液溶解介質為1.0 mol/L鹽酸溶液,Cr6+標準溶液溶解介質為水,因此本實驗采用加標法測定回收率驗證這兩種不同價態、不同溶解介質的鉻標準曲線對水溶肥料鉻形態含量測定結果的適用性。在空白樣品(水)中加入Cr3+和Cr6+混合標準溶液,分別采用這兩種不同溶解介質、不同價態的標準曲線,按所建立方法考察水溶性總鉻和水溶態Cr3+的加標回收率,具體方法和結果見表2。
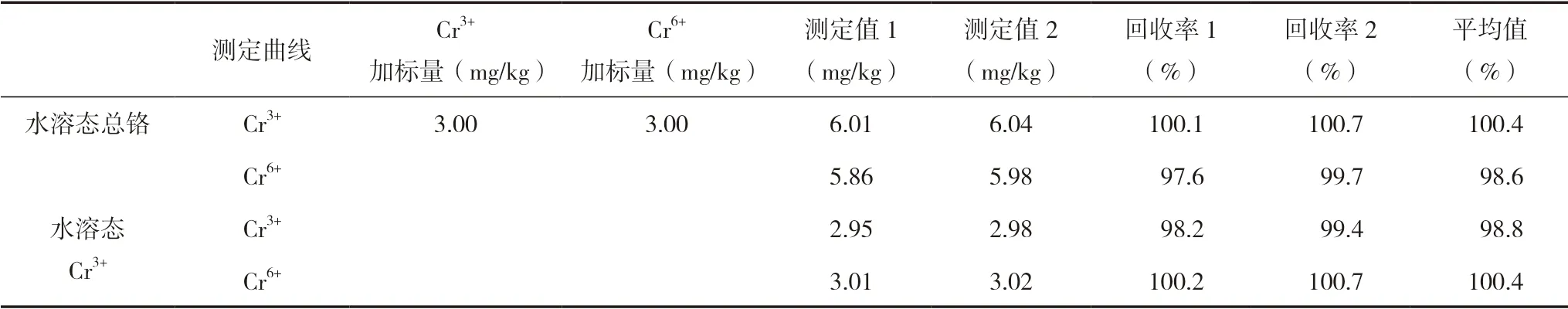
表2 不同鉻標準曲線測定回收率
實驗數據表明,水溶態總鉻、Cr3+含量采用這兩種不同溶解介質、不同價態的標準曲線測定,回收率兩次平行測定結果一致性較好,且兩次測定平均值均在98.6%~100.4%范圍內。說明不同鉻標準曲線對測定結果無影響,且可以準確定量測定。由于水溶肥料樣品采取水提取,因此本實驗選用Cr6+標準溶液進行工作曲線的配制。
2.3 鉻形態分離效果
2.3.1 試樣溶液上柱流速的選擇
實驗研究了試樣溶液在不同的上柱流速下樹脂吸附Cr6+的情況(圖3)。結果表明,試液上柱流速較低時,試液中Cr6+在樹脂空隙間停留時間較長,Cr6+在樹脂上交換吸附較完全,而流速過高會造成樹脂吸附不完全。從實驗數據看,上柱流速小于1.0 mL/min時,Cr6+在樹脂上吸附近100%。因此,本實驗試樣溶液上柱流速選擇 1.0 mL/min。
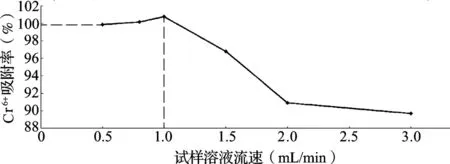
圖3 上柱流速對鉻形態分離的影響
2.3.2 鉻形態分離效果
為驗證采用安捷倫PAX 60 mg,3 mL陰離子交換柱分離Cr3+和Cr6+的適用性,選擇對實驗用水進行加標法測定回收率和吸附率。取2個空白樣品(水),分別加入Cr3+和Cr6+標準溶液,按所建立方法測定Cr3+和Cr6+的值,考察Cr3+加標回收率及Cr6+吸附率,具體加標方法和結果見表3。

表3 離子交換法提取鉻形態測定回收率/吸附率
實驗數據表明,在水溶液中加入Cr3+、Cr6+,Cr3+回收率為100.3%,Cr6+吸附率為98.7%。說明安捷倫PAX 60 mg,3 mL陰離子交換柱適用于Cr3+、Cr6+的分離。
2.4 回歸方程、線性范圍和檢出限
配制一系列濃度的Cr6+標準溶液,在所選擇的最佳實驗條件下進行測定,分別以鉻質量濃度 x(mg/L)為橫坐標、峰面積y作為縱坐標,得到回歸方程、線性范圍和相關系數,對Cr6+的空白溶液連續測定12次,依據國際純粹與應用化學聯合會(IUPAC)定義的檢出限表達式CL=KS/m計算出儀器檢出限。結果見表4。
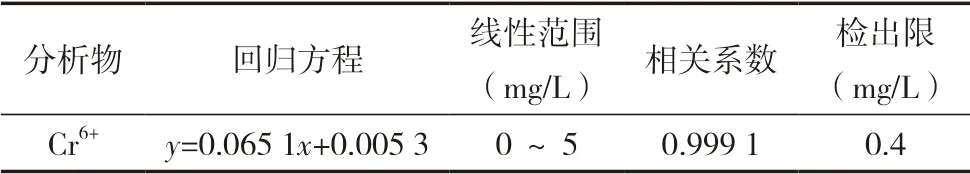
表4 Cr6+測定的回歸方程及線性范圍
2.5 方法精密度
為考察該方法的精密度,分別對3個樣品進行6次平行測定,水溶態總鉻測定值的相對標準偏差(RSD)為1.12%~3.13%,水溶態Cr3+測定值的RSD為1.93%~2.93%,結果分別見表5和 表6。

表5 水溶態總鉻精密度實驗結果

表6 水溶態Cr3+精密度實驗結果
2.6 方法準確度
通過加標回收實驗來評價本方法的準確性。在上述不同類型肥料試樣(S01~S03、S05、S06、S09、S10)中,加入定量的Cr6+、Cr3+標準溶液。經兩次平行測定,計算得出水溶態總鉻、Cr3+的回收率及Cr6+吸附率。
2.6.1 水溶態總鉻加標回收率、Cr6+吸附率實驗
選用不同pH、不同類型的肥料試樣,加入定量的Cr6+標準溶液。回收率及吸附率實驗結果如表7所示。總鉻回收率為99.0%~102.0%,Cr6+吸附率為97.0%~102.0%。回收率、吸附率結果達到分析準確度要求。同時也證明本實驗采取的離子交換法分離Cr3+和Cr6+,分離效果達到分析 要求。
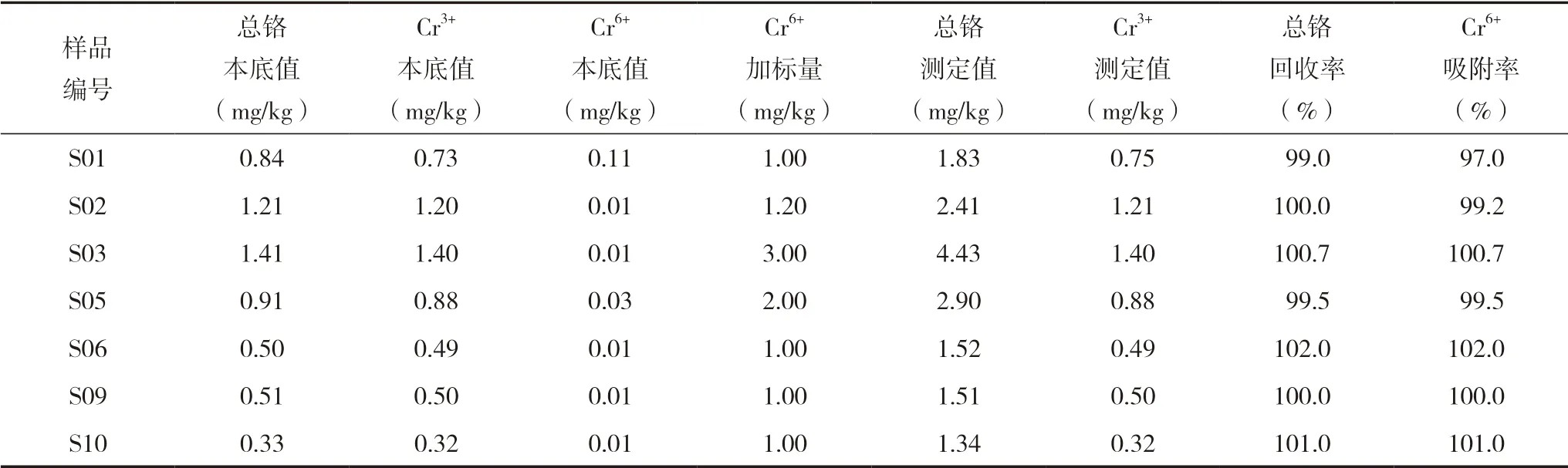
表7 水溶態總鉻、Cr6+回收率/吸附率實驗結果
2.6.2 水溶態Cr3+回收率實驗
選用不同pH、不同類型的肥料試樣,加入定量的Cr3+標準溶液。回收率實驗結果如表8所示。鉻回收率為98.8%~104.0%,回收率結果達到分析準確度要求。

表8 水溶態Cr3+回收率實驗結果
2.7 樣品基體對方法測定結果的影響
水溶肥料基體中往往存在大量銅、鐵、錳、鋅、鉬、硼、鈣、鎂、鉀等元素,以及氨基酸、腐植酸等有機物質。由于樣品基體成分含量高,而且變化不定等因素,很難配置與水溶肥料樣品溶液相似基體的標準溶液,因此采用標準加入法與標準曲線法比較來考察樣品中是否存在影響實驗結果的干擾物質。
本實驗選取含氨基酸、腐植酸、有機質、微量元素等基體成分的水溶肥料試樣進行標準加入法測定和標準曲線法測定,標準加入系列溶液見表9,結果見表10。
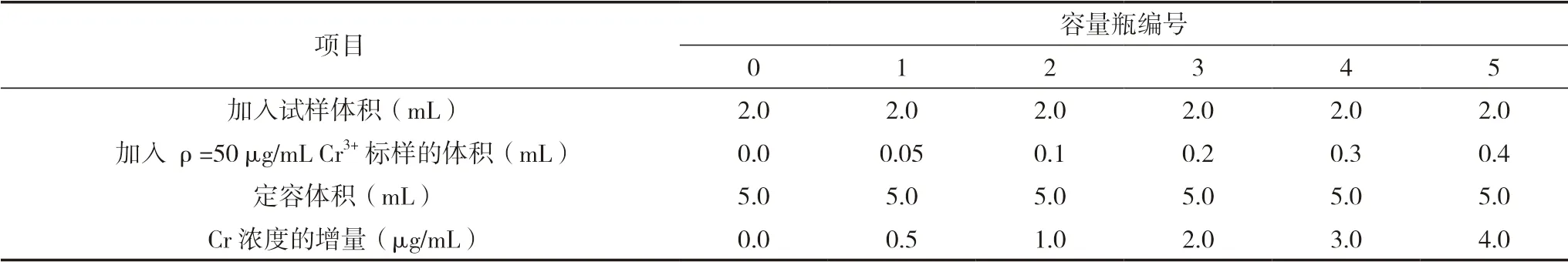
表9 標準加入系列溶液
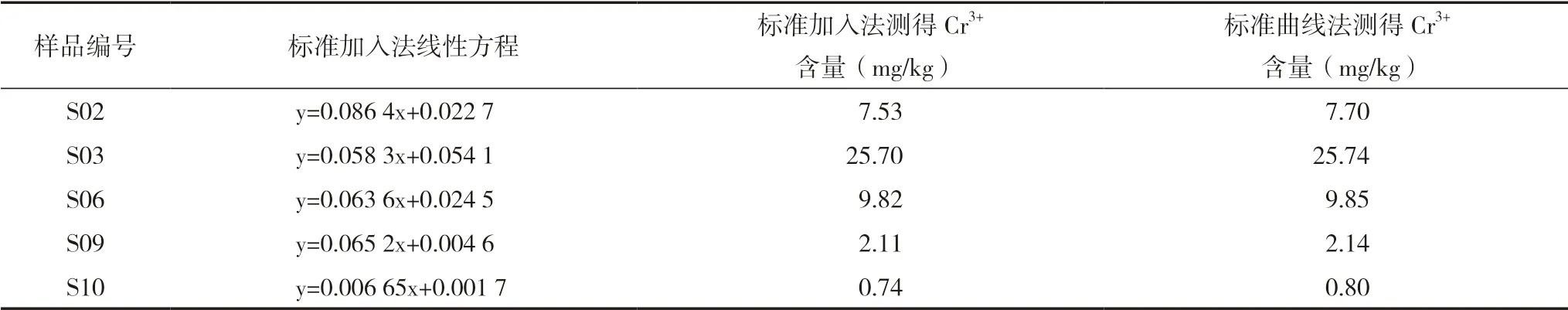
表10 標準加入法與標準曲線法Cr3+含量測定比較
表10實驗結果表明,采用標準加入法測定結果與標準曲線法測定結果符合性較好,從而證明樣品溶液中氨基酸、腐植酸、有機質、微量元素等基體成分對原子吸收分光光度法測定鉻含量無影響。
2.8 樣品待測液穩定性測試
為了考察試樣提取溶液的穩定性,重復測定之前測過并于4℃冰箱保存的試樣溶液中水溶態總鉻、Cr3+含量,結果分別見表11和表12。
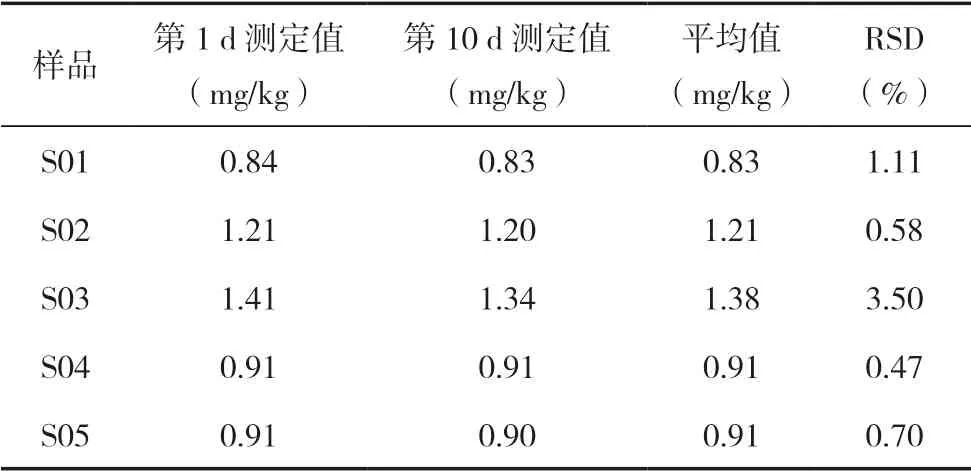
表11 水溶態總鉻穩定性實驗結果

續表

表12 水溶態Cr3+穩定性實驗結果
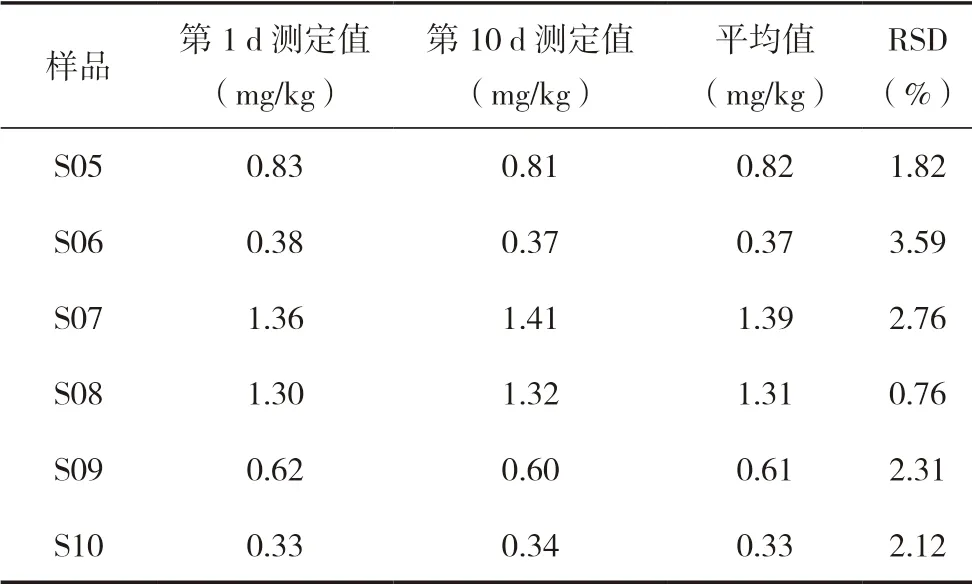
續表
表11和表12結果表明,經本實驗法提取的試樣溶液的水溶態總鉻測定值第1 d和第10 d的相對標準偏差(RSD)為0.58%~3.75%,水溶態Cr3+測定值第1 d和第10 d的RSD為0.76%~3.59%,說明試樣溶液保存在4℃冰箱,溶液的水溶態總鉻和Cr3+含量在10 d內較穩定。
2.9 樣品待測液酸化對測定結果的影響
待測液酸化處理可以防止金屬離子沉淀,因此吸取不同類型的肥料試樣水溶態總鉻、Cr3+待測溶液各4 mL,加入0.2 mL鹽酸進行酸化處理,處理后測定結果見表13。
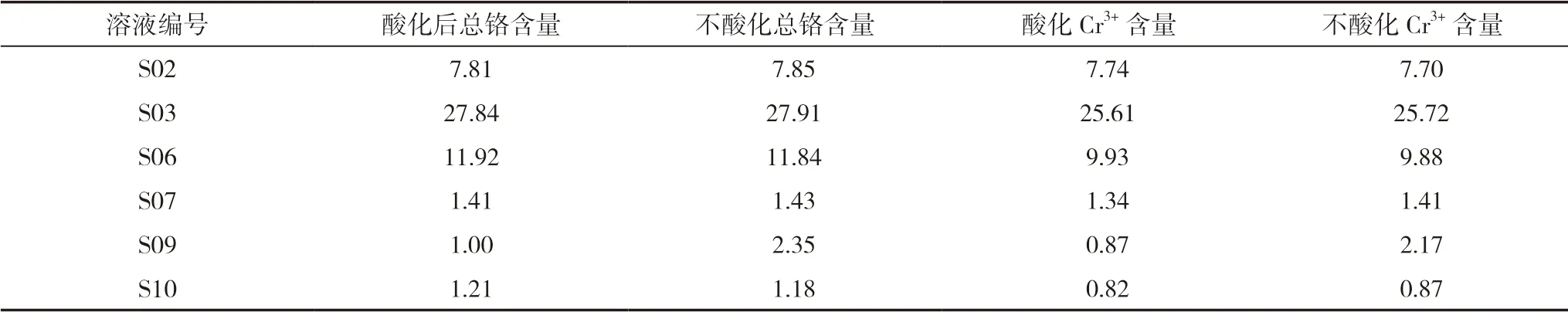
表13 水溶態總鉻、Cr3+酸化處理與未酸化處理測定值比較 (mg/kg)
實驗結果表明,除S09酸化后水溶態總鉻、Cr3+含量降低外,其余結果符合性較好。經分析,S09為腐植酸樣品,溶液酸化后引起腐植酸沉淀,導致Cr3+金屬元素一同被沉淀,造成結果偏低。因此,本實驗對樣品待測液不進行酸化處理。
3 結論
本實驗采用陰離子交換柱吸附Cr6+,使之與Cr3+分離,火焰原子吸收光譜法測定水溶肥料中水溶態Cr3+及總鉻含量,差減法測得Cr6+含量。建立了僅含Cr3+和Cr6+的水溶肥料中鉻形態的固相萃取-FAAS測定方法。研究結果表明,本實驗方法采用固相萃取分離水溶肥料中的Cr3+和Cr6+,Cr3+回收率和Cr6+吸附率分別為100.3%、98.7%,分離效果較好。使用FAAS法進行檢測,Cr3+的加標回收率在98.8%~104.0%之間,Cr6+吸附率在97.0%~102.0%之間,滿足檢測分析的需要,且方法操作簡便,準確度高,精密度好,適用于水溶肥料中Cr3+、Cr6+的檢測。
4 展望
雖然現在肥料中鉻的形態分析已經受到人們的重視,但系統的理論研究較缺乏,實際應用方面也很有限,所以選擇簡便、快速的分離富集技術和高靈敏度、高選擇性的檢測方法仍是今后的主要發展方向,應主要集中在提高方法的選擇性,盡可能地減少或免去樣品的預處理過程等方面。本實驗采用差減法測定Cr6+含量,如何將Cr6+從交換柱上洗脫并進行濃縮富集測定,如何防止分析過程中形態的改變、痕量污染或損失等方面也是今后肥料鉻形態分析中需要加倍關注和研究的重點。
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
