畜禽微生物耐藥組研究進展
2021-01-22 09:30:40馬濤陸唯李松勵樊霞
生物技術通報 2021年1期
馬濤 陸唯 李松勵 樊霞
(1. 中國農業科學院飼料研究所 農業農村部飼料生物技術重點實驗室,北京 100081;2. 中國農業科學院北京畜牧獸醫研究所,北京100193;3. 中國農業科學院農業質量標準與檢測技術研究所 國家飼料質量監督檢驗中心,北京 100081)
雖然抗生素自問世以來已經挽救了數百萬人的生命,但伴隨產生的抗生素耐藥性也對全球公眾健康造成了巨大威脅。如果不加以改善,預計到2050年,每年因抗生素耐藥性導致死亡的人數將從70萬上升至1000萬[1]。抗生素耐藥性問題產生的主要原因并非僅僅在于人類自身不合理地使用抗生素,還在于絕大部分(73%)抗生素用于給人類提供肉奶蛋等食品的養殖動物[2]。事實上,自20世紀50年代起,抗生素就應用于畜禽生產,起到了提高飼料轉化效率、促進生長或治療疾病/降低疾病發生率的作用[3]。尤其是2000年以來,非洲、亞洲和南美等中低收入國家的肉類產量分別增長了68%、64%和40%[2],這很大程度上得益于集約化生產體系全球化的擴展和普及,而抗生素的使用對于維持動物健康和提高生產力起到了至關重要的作用[4]。全球用于雞、豬和牛的抗生素總使用量將從2010年的約6.3×103t萬噸增加到2030年的約10.5×103t噸,增幅高達67%[5]。越來越多的證據證明抗生素在畜禽生產上的大量使用與人類抗生素耐藥性問題的加重存在緊密聯系[6-8],這是因為畜禽消化道或畜產品(如牛奶)微生物攜帶的抗生素耐藥基因(Antibiotic resistance gene,ARG)可以轉移到人類消化道微生物中[9]。基于此,2017年世界衛生組織呼吁其成員國減少獸用抗菌藥物的使用[10-11]。我國已經禁止在日糧中添加任何促生長的抗生素,然而動物疫病發生時還需要使用抗生素治療。因此,抗生素耐藥性是21世紀全球面臨的最緊迫的挑戰之一,對現代醫學和食品安全構成威脅。
為了降低畜禽生產環節ARG向人類的傳播,首先需要明確畜禽消化道或產品微生物攜帶哪些ARG。傳統上通過分離含有潛在ARG的細菌并提取其DNA,再通過PCR或者比較全基因組測序的方法明確該細菌含有哪些ARG[12]。但該方法的局限性在于僅能研究單一的、可體外培養的細菌。近年來,基于高通量測序技術來分析ARG的研究越來越受到青睞,因為該方法與傳統技術相比擴大了ARG的監測范圍[13-14]。例如,使用宏基因組學來確定某個環境微生物群落全部抗生素耐藥性基因—耐藥組(Resistome)[15-16]組成和相對豐度的方法已廣泛應用于人類腸道[17-18]、水源[19-21]、土壤[22-23]等微生物群落的研究中,畜禽消化道微生物耐藥組研究也開始受到關注。本文首先介紹了微生物耐藥組的研究方法,隨后對近年來畜禽消化道和乳中微生物耐藥組及其影響因素研究進展進行了總結和歸納,最后提出了未來研究方向,包括研究方法的標準化、耐藥組基因表達的研究等,旨在為控制畜禽養殖過程中ARG向人類的傳播提供思路。
1 微生物耐藥組的研究方法
1.1 基于序列的研究方法
該方法是利用生物信息學分析工具,將宏基因組或宏轉錄組測定的細菌基因組或轉錄組序列與ARG數據庫進行比對來實現。具體而言,首先向分析工具提供測定的DNA或RNA序列,分析工具隨后使用不同的搜索算法與ARG參考數據庫進行比對,最終確定該序列是否存在已知的ARG(圖1)。上述流程可利用商業公司提供的專有平臺來實現,或者從github及bitbucket等開源平臺上下載工具來完成分析。生物信息學分析工具雖然多種多樣,但按照接受輸入數據類型的不同,主要分為可分析組裝后序列和可分析原始序列兩大類[24]。需要指出的是,不論何種分析工具都不能對輸入序列數據質量進行控制,因此需要首先對原始序列或組裝后序列進行質量控制,再進行耐藥組分析。
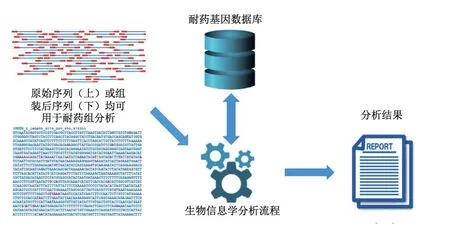
圖1 基于宏基因組測序的微生物耐藥組分析流程[25]
基于原始序列和基于組裝后序列的分析工具各有優缺點:使用可分析組裝后的序列的工具時,組裝軟件的差異可能會影響結果的可比性[26-27],這時由于序列組裝后,將輸入數據與ARG參考數據庫進行比較的最常見方法依賴于BLAST和隱馬爾可夫模型搜索[28]。基于BLAST的工具可以根據基因長度和相似性百分比的默認設置給出不同的輸出結果,如果設置太低或太高,都會對特異性產生負面影響。此外,基于組裝的方法對于計算機配置要求很高。盡管有這些缺點,但是基于組裝的方法可以分析ARG的遺傳背景[25]。另一方面,使用可分析原始序列的工具時,其將序列數據與ARG數據庫進行比對的方式較多,包括Bowtie2、BWA和KMA等[24]。該類分析工具的缺點是其比對時間相對于使用可分析組裝后序列的工具較長,但其優點在于能夠檢測出由于組裝不完整可能會被忽略掉的耐藥基因[24]。
除分析方法外,耐藥組預測準確性還取決于耐藥基因參考數據庫的完整性。耐藥基因參考數據庫可細分為專門用于檢測特定類別的耐藥基因(例如ARGO參考數據庫用于檢測耐β-內酰胺(beta-lactam)和萬古霉素(vancomycin)基因[29]),和用于檢測出DNA/氨基酸序列中理論上可能存在的耐藥基因(如ARDB[30]、ResFinder[31]、CARD[32]數據庫)。除了上述側重點不同,不同耐藥基因參考數據庫還具有其他重要特征。首先,耐藥基因參考數據庫中收錄條目的標準不同:如CARD中的條目必須已在科學文獻中發表[32],而在ResFinder對條目是否發布無嚴格的要求[31]。其次,條目的類型在參考數據庫之間也不同,大多數參考數據庫包括全部耐藥基因,只有少數參考數據庫也包括了染色體突變的耐藥基因(如MUBII-TB-DB[33]和PointFinder[34])。最后,耐藥基因參考數據庫在條目格式(fasta或json等)、下載權限,以及定期維護頻率方面也有所不同[25]。因此在選擇最佳的耐藥基因數據庫時需要對這些區別特征有充分的了解。
1.2 基于功能宏基因組學的研究方法
功能宏基因組學是一種多步驟的研究手段,其利用編碼混合微生物群落的分離DNA的表達文庫來轉化相關的宿主微生物。隨后可以從廣泛的目標表型中選擇對應的轉化克隆,如酶促、脂解或水解活性[35]。利用功能宏基因組學方法,結合分子生物學技術,篩選并構建抗生素耐藥基因庫,最后再利用生物信息學分析,確定新型的ARG[36-37]。應用功能宏基因組學的微生物耐藥組研究流程如圖2所示。
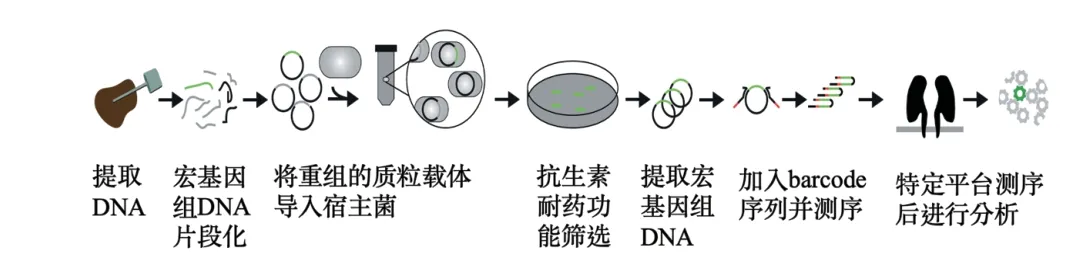
圖2 基于功能宏基因組學的微生物耐藥組分析流程[36]
宏基因組文庫構建的第一步是DNA提取,需要盡力保證完全提取樣品中的總DNA。此外,還需要得到足夠的片段以獲取表達所需的完整的目的基因或基因簇[38];隨后選擇適宜的載體(如質粒)和宿主菌(如大腸肝菌),利用提取的DNA構建宏基因組文庫,根據所用載體的不同,需要將DNA片段化,以確保載體連接和轉化效率,然后通過化學轉化法或電轉化法將重組的質粒載體導入宿主菌,實現宏基因組文庫的構建[39]。
文庫構建完成后,即可進行抗生素耐藥功能篩選,由于構建宏基因組文庫的宿主菌大多為革蘭氏陰性大腸桿菌,因此盡可能避免革蘭氏陰性菌不敏感抗生素(如糖肽類、大環內酯類、萬古霉素等),通常選擇在環境中檢出率較高的抗生素(如四環素類、磺胺類、大環內酯類和喹諾酮類)[39]。在進行抗生素耐藥性篩選時,一般使用抗生素的最小抑菌濃度,收集在此濃度下存活的宿主菌(陽性克隆),提取其DNA,通過PCR擴增并加入barcode序列[40];對DNA進行質檢后,構建測序文庫,用于高通量測序(如Nanopore或Pacbio等平臺),最后通過生物信息學分析手段,檢測所篩選的ARG,具體可通過poreFUME等集成式的分析流程分析[36],也可自行選擇并組合分析軟件進行分析[41]。
2 畜禽消化道微生物耐藥組分析
畜禽消化道微生物及其耐藥組的分析研究在近年開始受到關注[42]。畜禽消化道微生物耐藥基因既可能來源自母體,也可能通過與周圍環境中的耐藥性微生物(主要是細菌)直接接觸而獲得[43]。另一方面,消化道微生物具有廣泛而多樣的遺傳庫,可促進耐藥性在常駐共生菌種之間和內部的傳播[44]。從目前研究報道來看,四環素(Tetracycline)耐藥基因是畜禽(包括豬、雞、反芻動物)消化道微生物普遍存在的耐藥基因。
2.1 豬的消化道微生物耐藥組分析
已有研究證明未接觸抗生素的豬糞便中存在耐藥性基因[45-46],其中最主要的是四環素耐藥基因[47]。在沒有使用抗生素的情況下,豬場中經常發現的四環素耐藥基因包括tetO、tetW、tetM、tetX和tetQ,另外也有多個大環內酯類(Macrolide)抗性基因(ermG、ermF和ermB)[48];同樣,新生仔豬在未接觸抗生素的情況下糞便中也有ARG[49]。基于宏基因組測序結果,我國學者對國內4個大型豬場的豬糞耐藥組進行了分析發現,四環素、氨基糖苷(Aminoglycoside)和多藥(Multidrug)耐藥基因占全部耐藥基因相對豐度的近70%[50]。Munk等[51]對歐洲9個國家豬的糞便耐藥組進行了分析,結果表明不同國家豬糞便耐藥基因組成和比例相對一致,四環素耐藥基因相對豐度最高,超過了60%,其次是大環內酯、β-內酰胺和氨基糖苷類等基因。此外,共發現了33個核心ARG,包括VanG、tetC、blaACI和cfxA等。最近有研究表明,四環素、β-內酰胺和多藥耐藥類耐藥基因表達量與大腸埃希氏菌(Escherichia)和普雷沃氏菌(Prevotella)相對豐度存在正相關[52],說明這些菌可能是上述耐藥基因的主要宿主,為通過調控微生物組成降低豬生產中耐藥基因向環境中的傳播提供了依據。
2.2 家禽的消化道微生物耐藥組分析
Munk等[51]對歐洲9個國家家禽的糞便耐藥組進行了分析,其中四環素、大環內酯和氨基糖苷類等基因為主,但三者相對豐度較接近,與豬糞便ARG中四環素耐藥基因占絕對優勢有很大的區別;此外共發現了49個核心ARG,包括strAB、sul2、blaTEM和tetA基因等。金霉素是家禽和豬生產中最常用的抗菌藥物之一[53]。我國學者研究了使用金霉素對肉雞糞便耐藥組的影響,結果發現使用治療劑量的金霉素促進了糞便四環素耐藥基因tetA和tetW的豐度,并抑制了多藥耐藥基因mdtA、mdtC、mdtK、ompR和TolC的豐度[54]。此外,治療劑量的金霉素導致變形菌門(Proteobacteria)相對豐度降低,主要是由于其降低了該菌門中大腸埃希菌/志賀氏菌屬(Shigella)的相對豐度(從72%降至58%)。金霉素對大腸埃希菌的抑制作用是治療劑量組中多藥耐藥基因減少的主要原因,因為大腸埃希菌是多藥耐藥基因的主要宿主[54]。
2.3 反芻動物的消化道微生物耐藥組分析
作為反芻動物獨有的消化器官,瘤胃微生物耐藥組在近年來受到廣泛關注。Auffret等[55]比較研究了不同精粗比、品種、使用抗生素對肉牛瘤胃微生物耐藥組的影響,結果表明,肉牛瘤胃耐藥基因以大環內酯、氯霉素(chloramphenicol)、β-內酰胺和氨基糖苷為主;品種對于瘤胃耐藥組的影響不顯著;飼喂高粗料的肉牛瘤胃樣品中氯霉素和微霉素(microcin)耐藥基因占主導地位,而飼喂高精料的肉牛對氨基糖苷和鏈霉素的耐藥性更豐富。此外,高精料日糧還增加了變形菌門的相對豐度,其中包括許多動物和人畜共患病原體。Thomas等[56]研究發現飼糧中添加莫能菌素和泰樂菌素的肉牛瘤胃中檢測到了大環內酯類耐藥基因(ermF和ermG),在未添加抗生素的肉牛瘤胃中檢測到了氨基糖苷類耐藥基因(aadE和aph(3)-III)。上述研究結果表明牛瘤胃微生物普遍存在耐藥基因,與抗生素飼料添加劑的使用沒有必然聯系。目前關于羊瘤胃微生物耐藥組研究非常有限,Hitch等[57]在綿羊瘤胃中發現了30種不同的耐藥基因,以達托霉素(daptomycin)耐藥基因最為常見。
除瘤胃外,還有學者研究了使用抗生素對肉牛糞便微生物耐藥組的影響。飼糧中添加莫能菌素后,肉牛糞便中共檢測到了43種耐藥基因,主要為氨基糖苷、β-內酰胺、四環素和大環內酯-林可酰胺-鏈霉菌素 B(macrolide-lincosamide-streptogramin B)類,與沒有添加莫能菌素的肉牛相比,四環素和MLS等耐藥基因相對豐度并未降低[58]。北美經常在肉牛抵達飼養場后注射抗生素以控制呼吸道疾病,土拉霉素是一種用于預防與呼吸道疾病相關的抗生素[59-61],給新進牛只皮下注射土拉霉素后,分別在注射后的第1天和第11天比較對照組和注射組微生物和耐藥組差異,結果發現兩組之間的微生物組和耐藥組成并沒有顯著差異。這些結果表明,與普通的代謝型抗微生物藥物治療相比,向飼養場的過渡以及飲食、地理環境的變化可能對飼養場牛的糞便微生物耐藥組的影響更大[62]。此外,最近一項研究發現,飼養過程中使用抗生素的牛場糞便中的耐藥基因要高于不使用抗生素牛場糞便中的耐藥基因豐度,其中育肥牛的四環素和大環內酯類-林可酰胺-鏈霉菌素B類耐藥性比奶牛糞便中豐富,而β-內酰胺類在奶牛糞便中更豐富,但過程分析(Procrustes analysis)發現微生物群落與耐藥組缺乏一致性[63]。最近有學者研究了使用抗生素對犢牛糞便微生物耐藥組的長效作用影響。將42只小牛犢隨機分為3組,第一組在5 d內每天口服兩次1 g土霉素(高土霉素),第二組在7周內每天接受100-200 μg的土霉素(低土霉素),第三組未接受土霉素(對照組),通過宏基因組測序分析了對腸道菌群和耐藥基因豐度的時間影響。結果表明3種耐藥基因(tetM、floR和mel)的相對豐度在對照組和抗生素之間存在顯著差異。通過qPCR驗證宏基因組測序結果發現高土霉素組在第28-35天的tetM豐度達到峰值,而在低土霉素組中未發現任何耐藥基因豐度的增加[64]。除使用抗生素外,最近也有研究表明飼糧中添加釀酒酵母對肉牛糞便中的耐藥組無顯著影響[65],該研究結果表明四環素耐藥基因的相對豐度最高,包括tetQ、tetO、tetW和tet32;大環內酯-林可酰胺-鏈霉菌素 B的mefA基因相對豐度最高,其次是ermq、mphb和lunc;氨基糖苷類耐藥基因中,相對豐度最高的是ant9,其次是cfx。上述研究表明除使用抗生素或益生菌外,其他因素(例如農場的位置、牛源、管理模式等)都可能影響了糞便耐藥組,因此無法簡單地將糞便耐藥組歸咎于單一因素的影響。
3 乳中微生物耐藥組分析
據估計,美國至少有3%的人口消費未經巴氏消毒的生牛乳,且對于生牛乳需求持續增長。但是,食用原奶會導致食源性疾病,并且是含有可轉移的抗微生物耐藥基因(ARG)的細菌的來源。Liu等[66]比較分析了加利福尼亞州生牛乳和不同方式巴氏消毒后牛乳中微生物組及其耐藥組。結果表明剛采集的生牛乳和高溫瞬時巴氏消毒的牛乳中未檢測到任何耐藥基因,在室溫下放置24 h后,生牛乳中檢測到了49種耐藥基因,分別為多藥、氨基糖苷、β-內酰胺和四環素這4大類耐藥基因。對耐藥組的微生物宿主進行預測發現,9個已知菌科可能攜帶這些ARG,其中假單胞菌科(Pseudomonadaceae,36個)具有最多的獨特ARGs、其次是腸桿菌科(Enterobacteriaceae,28個)、耶爾森菌科(Yersiniaceae,14個)和莫拉菌科(Moraxellaceae,8個)。最近研究發現坦桑尼亞北部地區居民攜帶的耐藥性大腸桿菌可能來源自生牛奶[67];巴西的一項研究表明,從水牛奶中分離出的凝固酶陰性葡萄球菌可能是潛在的耐藥基因如耐甲氧西林基因(mecA)的宿主[68]。上述研究表明,耐藥基因經牛奶向人體中的傳播對人類健康造成的潛在危害可能比特定的病原體更為普遍,因此需要特別關注消費未經巴氏消毒奶制品的低收入國家[69-70]的微生物食品安全風險。
4 未來研究方向
全球范圍內對于畜禽消化道及其產品微生物耐藥組分析研究已經取得了一定的進展,主要體現在初步明確了上述耐藥組的基因組成和相對豐度。然而目前微生物耐藥組分析還存在著一定的局限性,從分析技術方法和研究思路方面還需要進一步拓展挖掘。
首先,目前科學文獻中至少公開了近50種可用于分析耐藥組的資源[71-72],既包括了可以嵌入用戶自己的生物信息學流程的基礎ARG參考數據庫,也包括了自帶參考數據庫且集成搜索工具的全套分析工具。由于這些分析工具的功能差異很大,因此需要考慮使用不同工具獲得的結果的可比性[73]。
其次,耐藥組依賴于細菌組成,而后者的分析受限于已知的微生物數據庫,鑒于細菌數據庫的局限性,目前尚有大量無法鑒定的微生物,因此已知的ARG可能僅代表實際抗菌素耐藥菌種群的一小部分。可以合理假設,隨著細菌基因組測序和功能宏基因組學的爆炸式增長以及數據庫的不斷完善,將鑒定出許多以前功能未知且無法單獨通過序列識別的新型ARG[24]。
第三,細菌產生耐藥性的一個重要原因是能夠通過質粒(Plasmid)之類的可移動遺傳元件(Mobile genetic element,MGE)獲得耐藥基因的能力[74]。這是由于細菌之間存在廣泛的水平基因轉移(Horizontal gene transfer,HGT)[75-76]。質 粒 介導的耐藥基因包括qnrA、blaCTX-M和mcr-1等[77]已追溯到了其環境和動物起源,然而目前針對畜禽微生物質粒等MGE攜帶的耐藥組研究仍然非常有限。已有研究表明,質粒是牛瘤胃細菌群體中大量存在的MGE之一[78],未來可基于ACLAME[79]和PlasmidFinder[80]等參考數據庫進一步明確畜禽消化道或產品中MGE攜帶的耐藥組,為限制耐藥基因通過HGT在細菌之間傳播提供依據。
第四,目前畜禽微生物耐藥組研究主要基于宏基因組短鏈[55-56]或長鏈測序[81]等技術,基因表達能夠更好地評估生物生態系統內功能活性[82],但耐藥組的表達及其影響因素尚不清楚。最近已有研究利用宏基因組學和宏轉錄組評估了廢水處理廠的耐藥組,并揭示了工廠位置不僅影響耐藥組組成,而且影響具體耐藥基因的表達[83]。最近也有基于宏轉錄組學研究分析了雞和豬腸道的耐藥組,但其樣本數理有限(每種動物只有6個樣品)[84],因此未來需要更多基于宏轉錄組學的研究,充分了解耐藥基因的表達情況,從而有針對性地對其進行調控。
最后,目前耐藥組研究的最大局限在于沒有對耐藥基因的功能進行驗證,通過基因注釋獲得的耐藥基因未必具有真正的耐藥活性,因此一定程度上限制了將ARG微生物標記物用于新興耐藥機制的臨床應用。對于畜禽等養殖動物腸道等環境中新型ARG的存在以及上述ARG的耐藥和傳播機制有待于系統地研究和進一步驗證[85]。
5 總結
耐藥組分析表明畜禽消化道及其產品微生物存在豐富的耐藥性基因,這些耐藥性基因的存在可能與抗生素的使用沒有直接聯系,但其組成和相對豐度可能受到抗生素、益生菌或其他飼料添加劑的影響。未來需要進一步完善耐藥組分析手段和技術,探明耐藥組的表達以及質粒攜帶的耐藥組,從而獲得完整全面的畜禽消化道微生物耐藥組分析結果。這將有助于充分評價動物養殖過程中耐藥基因向人類傳播的可能性,并通過有效調控途徑降低耐藥性基因對人類健康和環境的威脅。
猜你喜歡
保健醫苑(2022年5期)2022-06-10 07:46:38
昆明醫科大學學報(2022年1期)2022-02-28 07:43:40
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
財經(2017年2期)2017-03-10 14:35:35
山東工業技術(2016年15期)2016-12-01 05:31:22
財經(2016年15期)2016-06-03 07:38:02
財經(2016年3期)2016-03-07 07:44:46
財經(2016年6期)2016-02-24 07:41:51
中國當代醫藥(2015年17期)2015-03-01 02:03:58
