PAL探針中間體3-氧代-6-庚炔酸酯的合成方法改進
2021-01-28 06:16:52何冠濤方成喬羅旭娜蘇金龍林漢森
合成化學 2021年1期
何冠濤, 方成喬, 羅旭娜, 蘇金龍, 饒 雨, 林漢森
(廣東藥科大學 藥學院,廣東 廣州 510006)
光親和標記(photo-affinity labeling, PAL)自1962年首次報道以來[1],迅速成為了藥物與靶蛋白研究之間的重要橋梁,不僅可以指導先導物的篩選,更能夠實現高效靈敏的藥物受體識別[2]。PAL探針是目標化合物鏈接光反應性基團(photo-reactive group)以及報告基團(reporter group)的衍生物[3],其中雙吖丙啶類PAL探針的合成及其靶標識別研究已有許多相關報道[4-7]。基于PAL探針的設計原則[8],李正球等[9-10]報道了一種具有簡約結構特點的端炔基雙吖丙啶類PAL的合成(Scheme 1),同時證明了此類PAL探針具有良好的靶標識別性能,目前國內外已有各種靶蛋白研究采用此方法來合成PAL探針[11-16]。
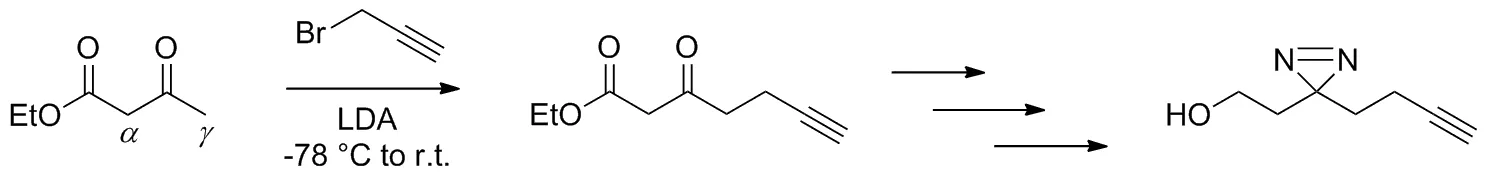
Scheme 1
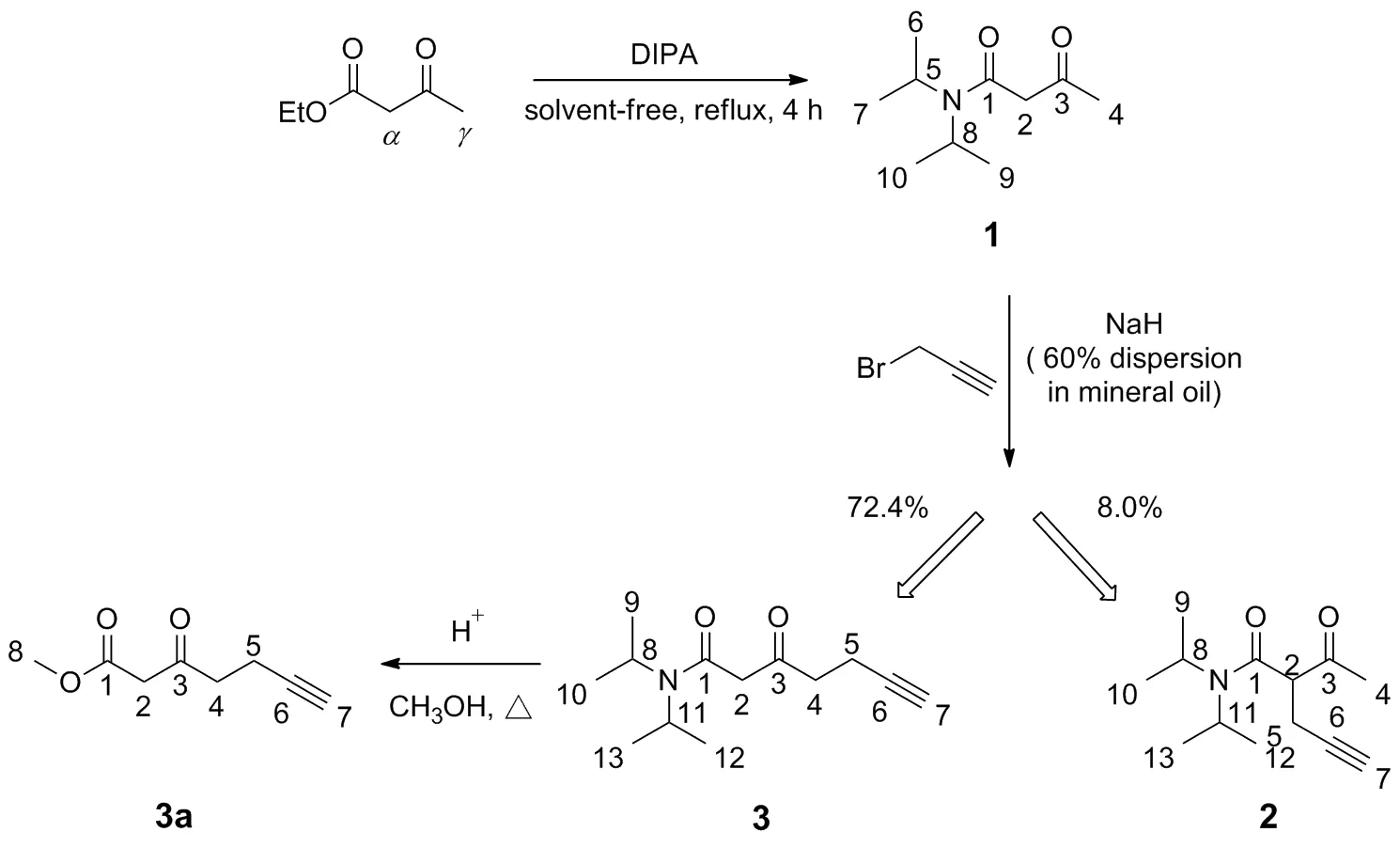
Scheme 2
在研究該中間體3-氧代-6-庚炔酸酯的合成方法時,發現上述制備方法略有不足:一方面,使用二異丙基氨基鋰(LDA)直接與原料中的γ-H作用形成γ碳負離子,然而LDA的使用條件苛刻,反應需要在極低溫(-78 ℃)、無水、無氧和氮氣保護下進行,且后處理復雜,對儀器設備要求較高。另一方面,乙酰乙酸乙酯具有更加活潑的α-H,可與LDA作用形成α碳負離子,因此上述方法在原料與溴丙炔的親核取代中,往往會生成大量的α-取代物和α,γ-二取代物,而γ-取代物3-氧代-6-庚炔酸乙酯的實際收率僅有5%~15%,且重現性較差,難以達到預定的效果。為此,改進了3-氧代-6-庚炔酸酯的合成工藝(Scheme 2):利用二異丙胺氨解乙酰乙酸乙酯生成N,N-二異丙基乙酰基乙酰胺1,減弱α-H酸性,增大α-位空間位阻。之后再與3-溴丙炔進行親核取代,經硅膠柱層析分離得到α-取代物2與γ-取代物3,兩者比例為1/9,反應的區域選擇性以及γ-位目標取代物的收率均有所提高。3再經水解與酯化一鍋法反應得到3-氧代-6-庚炔酸甲酯3a。優化后的工藝操作簡便、后處理簡單、反應條件溫和,目標產物總收率可達43.3%。
1 實驗部分
1.1 儀器與試劑
Bruker AVANCEⅢ 600 MHz型超導核磁共振儀(CDCl3為溶劑,TMS為內標);6210 ESI-TOF型高分辨質譜儀。
所用試劑均為分析純。
1.2 合成
(1)N,N-二異丙基乙酰基乙酰胺(1)的合成
稱取乙酰乙酸乙酯2.3 g(18.0 mmol)、二異丙胺3.6 g(36.0 mmol),85 ℃反應4 h后冷卻至室溫,加水30 mL,攪拌,二氯甲烷(3×30 mL)萃取,飽和氯化鈉水溶液(50 mL)洗滌,無水硫酸鈉干燥,減壓濃縮得深黃色油狀液體13.10 g,收率93.0%;1H NMR(CDCl3, 600 MHz)δ: 3.70~3.63(m, 2H), 3.33(s, 2H), 2.08(s, 3H), 1.23~1.22(d,J=6.7 Hz, 6H), 1.03~1.02(d,J=6.7 Hz, 6H);13C NMR(CDCl3, 151 MHz)δ: 202.74, 165.25, 52.01, 45.77, 29.73, 20.18; HR-MS(ESI-TOF)m/z: Calcd for C10H19NO2{[M+H]+}186.1494, found 186.1489。
(2)α-取代物(2)和γ-取代物(3)的合成
稱取化合物13.1 g(16.7 mmol),氫化鈉0.8g(33.4 mmol)溶解于20 mL四氫呋喃中,冰水浴下攪拌0.5 h后,緩慢滴加3-溴丙炔2.0 g(16.7 mmol),低溫反應2 h(TLC監測)。加水40 mL和乙酸乙酯40 mL,攪拌,分液,水層用乙酸乙酯(2×30 mL)萃取。合并有機層,飽和氯化鈉水溶液(50 mL)洗滌,無水硫酸鈉干燥,減壓濃縮,殘余經硅膠柱層析[洗脫劑:V(乙酸乙酯)/V(石油醚)=1/5]純化得α-取代物2和γ-取代物3。
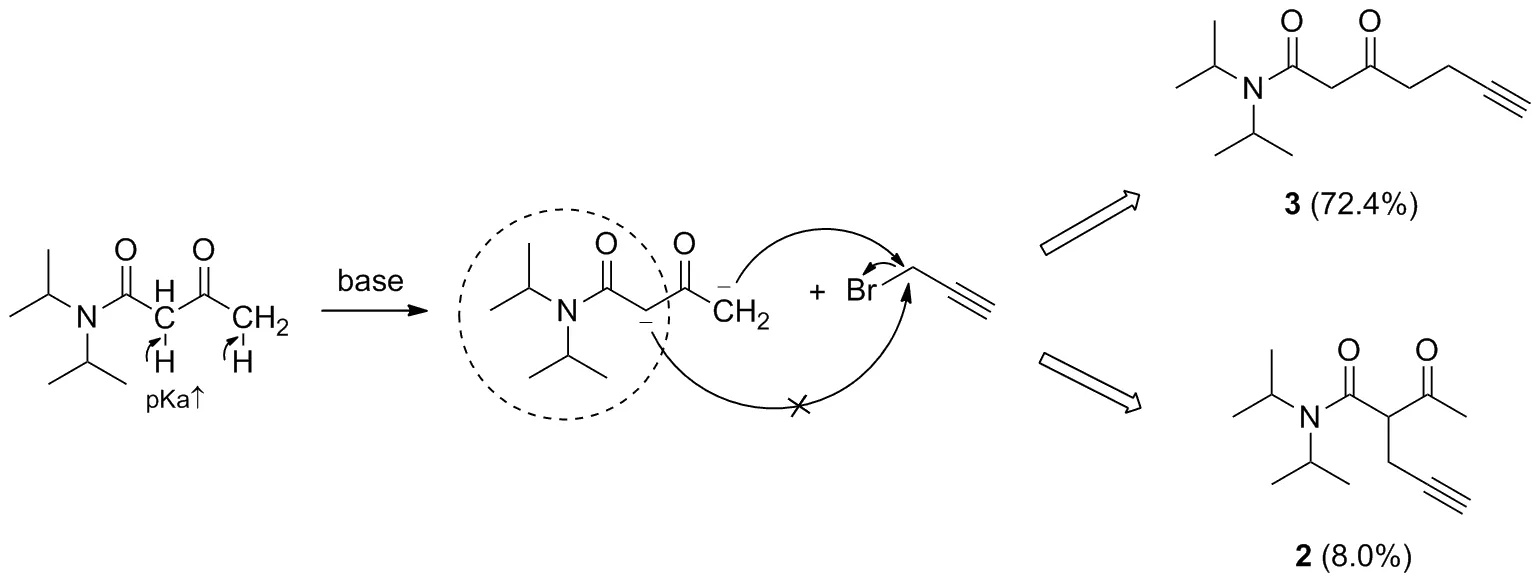
Scheme 3
α-取代物2: 黃色油狀液體0.3 g,收率 8.0%;1H NMR(CDCl3, 600 MHz)δ: 4.24~4.17(m, 2H, 8,11-H), 3.78~3.75(t,J=7.5 Hz, 1H, 2-H), 2.84~2.67(m, 2H, 5-H), 2.16(s, 3H, 4-H), 2.00~1.99(t,J=2.7 Hz, 1H, 7-H), 1.24~1.23(d,J=6.7 Hz, 6H, 12,13-H), 1.21~1.20(d,J=6.7 Hz, 6H, 9,10-H);13C NMR(CDCl3, 151 MHz)δ: 202.88, 166.76, 81.20, 70.53, 57.60, 46.62, 27.07, 20.73, 20.25, 18.67; HR-MS(ESI-TOF)m/z: Calcd for C13H21NO2{[M+H]+}224.1650, found 224.1645。
γ-取代物3: 黃色油狀液體2.7 g,收率 72.4%;1H NMR(CDCl3, 600 MHz)δ: 3.83~3.76(m, 2H, 8,11-H), 3.48(s, 2H, 2-H), 2.80~2.77(t,J=7.2 Hz, 2H, 4-H), 2.45~2.42(td,J=7.2, 2.7 Hz, 2H, 5-H), 1.91~1.90(t,J=2.7 Hz, 1H, 7-H), 1.36~1.35(d,J=6.7 Hz, 6H, 12,13-H), 1.16~1.15(d,J=6.7 Hz, 6H, 9,10-H);13C NMR(CDCl3, 151 MHz)δ: 202.93, 165.26, 83.00, 68.84, 51.33, 46.18, 41.47, 20.50, 12.98; HR-MS(ESI-TOF)m/z: Calcd for C13H21NO2{[M+H]+}224.1650, found 224.1645。
(3) 3-氧代-6-庚炔酸甲酯(3a)的合成
在裝有化合物32.7 g(12.1 mmol)的反應瓶中加入無水甲醇15 mL,油浴加熱升溫,攪拌下緩慢滴加80%硫酸15 mL,回流反應10 h。反應液緩慢倒入冷水50 mL中,攪拌,二氯甲烷(3×30 mL)萃取,依次用飽和碳酸氫鈉水溶液(50 mL)和飽和氯化鈉水溶液(50 mL)洗滌,無水硫酸鈉干燥,減壓濃縮,得淺黃色油狀液體3a1.3 g,收率64.3%;1H NMR(CDCl3, 600 MHz)δ: 3.71(s, 3H), 3.46(s, 2H), 2.79~2.77(t,J=7.2 Hz, 2H), 2.46~2.43(td,J=7.2, 2.7 Hz, 2H), 1.94~1.93(t,J=2.7 Hz, 1H);13C NMR(CDCl3, 151 MHz)δ: 200.55, 169.76, 83.97, 68.24, 51.92, 48.99, 42.58, 12.83; HR-MS(ESI-TOF)m/z: Calcd for C8H10O3{[M+H]+}155.0708, found 155.0703。
2 結果與討論
2.1 γ-取代物3的合成方法改進與工藝優化
通過二異丙胺氨解乙酰乙酸乙酯生成N,N-二異丙基乙酰基乙酰胺1后,反應起始物的α-位附近被引入一較大的供電子位阻基團,因此α-H的酸性減弱,同時α-位附近的空間位阻效應增大,阻礙了α碳負離子的形成及其對3-溴丙炔的進攻(Scheme 3)。改進后的親核取代反應具有更好的區域選擇性,α-取代物大大減少,γ-取代物明顯增多,兩者比例可達1/9,提高了后續合成3-氧代-6-庚炔酸酯的效率。
親核取代中選擇不同的堿對反應的影響也較大,一般的選擇有:LDA、正丁基鋰、氫化鈉、甲醇鈉、乙醇鈉、叔丁醇鉀。考慮到使用LDA和正丁基鋰所附帶的危險性,我們考察了氫化鈉、甲醇鈉、乙醇鈉、叔丁醇鉀4種相對較穩定的堿的催化效果,用量均為2 eq.。結果顯示,使用叔丁醇鉀時,1尚未完全轉化,α-取代物與γ-取代物比例約2/9,反應或需要更長的時間。而選擇使用氫化鈉時,1可完全反應,TLC顯示原料點消失,經柱層析純化后γ-取代物收率最佳,兩種取代物比例達到1/9。使用甲醇鈉、乙醇鈉時可見原料點基本消失,兩種取代物比例分別約為2/7和1/5,對比使用氫化鈉,反應生成的雜質斑點更加明顯。相比LDA,使用氫化鈉的反應體系更加溫和、操作簡便、后處理簡單,因此選擇氫化鈉代替LDA催化親核取代反應。
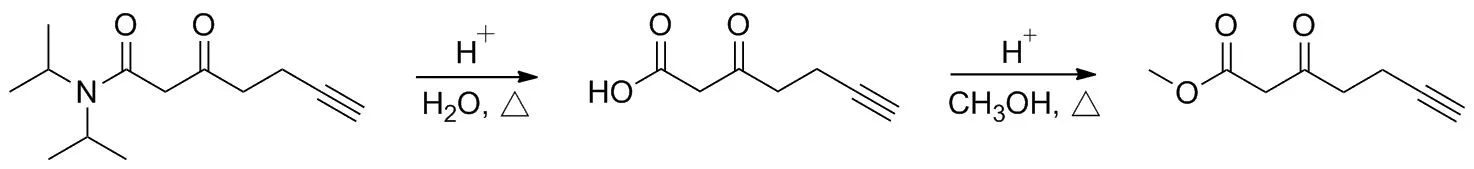
Scheme 4
2.2 3-氧代-6-庚炔酸甲酯3a的合成工藝優化
使用一鍋法合成3-氧代-6-庚炔酸甲酯3a,反應首先由γ-取代物3水解脫去位阻基團得到β-羰基羧酸,緊接著與甲醇酯化生成目標化合物(Scheme 4)。酰胺鍵的水解活性較低,需要在高溫以及濃酸或濃堿催化下長時間反應,但過高的反應溫度可能會導致水解物β-羰基羧酸發生脫羧,因此本研究選擇在100 ℃下使用濃硫酸催化水解以及進一步的酯化。在實際操作中,直接使用市售98%濃硫酸往往容易造成嚴重的碳化現象,導致后處理困難,影響收率。在考察使用不同濃度的濃硫酸(70%~90%)后發現,隨著硫酸濃度的升高,水解與酯化反應更加徹底,而在80%濃硫酸催化下效果最佳,收率約64%,同時避免了原料與產物被碳化。
以乙酰乙酸乙酯為原料,通過氨解引入N,N-二異丙基成酰胺以添加空間位阻效應后進行親核取代,得到α-和γ-兩種取代物比例為1/9,提高了反應的區域選擇性,且γ-取代物的收率達到72.4%,對比原路線中5%~15%的實際收率有所提升。γ-取代物再經水解與酯化一鍋法反應成功地合成了目標化合物3-氧代-6-庚炔酸甲酯,3步反應總收率43.3%。改進優化后的3-氧代-6-庚炔酸酯合成方法具有成本低、操作處理簡單、反應條件溫和等優點。
