一步法構建萊茵衣藻葉綠體表達載體
2021-03-04 07:40:32劉國興胡玉立徐丹丹
現代農業研究 2021年3期
劉國興,胡玉立,徐丹丹,徐 松
(國藥集團動物保健股份有限公司 湖北,武漢 430075)
萊茵衣藻是一種生活在淡水里的單細胞真核綠藻,其葉綠體表達的外源蛋白具有安全性好、培養周期短、遺傳穩定、容易擴大培養,并且成本低廉等優點[1,2,3]。近些年來,多種具有應用前景的蛋白在萊茵衣藻葉綠體中成功表達[4]。而一般構建衣藻葉綠體表達載體需要通過兩步克隆來完成:首先將外源基因插入克隆載體的衣藻來源的5’和3’非翻譯區(UTR)多克隆位點間,再將5’UTR-外源基因-3’UTR的表達盒酶切下,通過同源重組構建至表達載體[5,6]。為了簡化衣藻葉綠體表達載體的構建步驟,本研究構建了一種T載體pBTNK,通過一步TA克隆可將待表達的外源基因插入衣藻來源5’和3’UTR 之間,藍白斑篩選獲得陽性轉化子后,重組質粒可直接用于轉化植物,因此可簡化衣藻葉綠體中表達載體構建流程。
1 材料和方法
1.1 材料
1.1.1 藻株、菌株和質粒 萊茵衣藻Chlamydomonas reinhardtii137c (mt)、質粒 pCX16、pCX13、p228 由華中農業大學陳杏洲博士惠贈;大腸桿菌DH10β,質粒pShuttle、pFAST-EGFP、pT-BASIC由湖北大學蔣思婧博士惠贈。
1.1.2 酶、主要試劑和設備 BfuI 購自Thermo Scientific,LATaq酶、SolutionI 連接酶、T4DNA 聚合酶、質粒抽提試劑盒、MiniBEST Agarose Gel DNA Extraction Kit Ver.4.0、其他限制性內切酶,購自 Takara 公司,X-gal、dNTP、硫酸卡那霉素(kana)、氨芐青霉素購自北京九州同業生物科技公司,壯觀霉素購自sigma公司,PCR引物(表1)由上海捷瑞生物工程有限公司合成,Biolistic PDS-1000/He基因槍購于Bio-Rad。
1.2 方法
1.2.1 DNA 克隆 質粒提取參照OMEGA bio-tek 試劑盒說明書進行,DNA片段克隆、重組質粒鑒定等分子克隆操作參照Sambrook提供的方法進行[7]。
1.2.2 萊茵衣藻葉綠體轉化 根據文獻[8]描述的方法并適當調整后,通過基因槍法將基因導入葉綠體,即3μg載體質粒pTBNK-EGFP(1μg/μl)和2μg 抗性質粒p228(1 μg/μl)包被50μl(60 mg/mL)直徑為0.6μm金粉子彈,按壓力1100psi、真空度28英寸汞柱、目標距離6cm轟擊衣藻,將轟擊后的衣藻在TAP平板上暗培養15~20h后刮下,涂布于5皿含100μg/mL壯觀霉素抗性TAP平板,培養兩周。
1.2.3 葉綠體總DNA提取 衣藻葉綠體總DNA的抽提采用CTAB法[9]。
1.2.4 萊茵衣藻的培養 將萊茵衣藻培養在TAP 液體培養基或含1%瓊脂糖的固體平板上,在22℃~24℃、連續光照(約100 μE/m2.sec-1)條件下培養[10]。
2 結果
2.1 衣藻葉綠體表達載體pTBNK的構建
以質粒pCX16為模板,P1和P2為引物,LATaq酶PCR擴增5.7kb 同源臂上的2.0 kb 片段,回收PCR 產物后,用BamHI和SphI雙酶切并回收該2.0 kb片段,然后將酶切的片段插入pCX16 載體的BamHI 和SphI 酶切位點間,在pCX16 載體上引入NotI 酶切位點得到載體pCXN;EcoRI和SphI 雙酶切載體pCXN,回收pCXN 載體骨架5.7 kb 片段,并用T4DNA 聚合酶補平末端,將末端補平片段插入pT-BASIC 的EcoRV 酶切位點得到載體pTBN;以質粒pShuttle 為模板、P3 和 P4 為引物,LATaq酶 PCR 擴增 1.2 kb kana 抗性基因片段,將該片段回收后,BglⅡ和HindⅢ雙酶切該片段,然后將酶切后的片段插入pCX13 載體的BglⅡ和HindⅢ酶切位點間獲得載體pCX13-kana;用BamHI 將 pCX13-kana 載體上 5’UTR-BfuI-kana-BfuILacO 部分序列-3’UTR 表達盒酶切下,T4 DNA 聚合酶補平末端,然后和NotI 酶切、T4DNA 聚合酶補平末端的pTBN 載體片段用SolutionI 連接酶進行連結,獲得衣藻葉綠體定向表達T載體pTBNK(圖1)。
2.2 一步克隆構建衣藻葉綠體表達載體pTBNK-EGFP
以 pFAST-EGFP 為模板,P5 和 P6 為引物,LAtaqPCR擴增 EGFP 基因并回收該產物,BfuI 酶切 pTBNK 載體,連接后轉化至大腸桿菌DH10β 感受態細胞,涂布于含氨芐青霉素(50μg/ml)和X-gal(30μg/ml)的LB固體平板上,避光培養12~14h 后觀察,挑顯藍色的菌落(圖2)進行PCR鑒定(圖3)。經鑒定,陽性克隆達100%,表明重組衣藻表達載體pTBNK-EGFP構建成功。
2.3 表達載體pTBNK-EGFP導入衣藻葉綠體
pTBNK-EGFP 經基因槍法轉化萊茵衣藻葉綠體后,經100μg/mL壯觀霉素抗性篩選,共長出4個綠色單藻落,抽提野生型的葉綠體總DNA和第八次傳代的轉基因衣藻的葉綠體總 DNA 作為模板,以 P5 和 P6 為引物,PCR 擴增EGFP片段,結果顯示四個轉基因衣藻DNA樣本中均可以擴增出目的條帶而野生型DNA樣本無法成功擴增目的條帶(圖4),表明EGFP成功插入萊茵衣藻葉綠體。
3 討論
我們設計在載體克隆位點引入BfuI 酶切位點,該酶的特點是特異性識別5’-gtatcc-3’序列,并在該序列下游任意6 個堿基處對DNA 進行切割,切割后形成粘性末端“T”,而LAtaqDNA 聚合酶擴增的目的片段帶“A”尾。這樣,可通過TA克隆將目的片段克隆到pTBNK。
基于LacO重構原理[11],在PCR 擴增目的片段的下游引物5’增加7bp的部分LacO序列(5’-CACAATT-3’),載體骨架上攜帶 13bp 的部分LacO序列(3’-AATTGTGAGCGCT-5’),載體片段和目的片段連接后,轉化到lac+的菌株感受態細胞,涂布于含X-gal的平板,完整的LacO序列會競爭性的吸附宿主菌表達的LacI 蛋白,使宿主菌的乳糖啟動子控制的β-半乳糖苷酶基因表達解抑制,表達β-半乳糖苷酶,分解X-gal使菌落呈現藍色。因此,只有目的基因閱讀框正確插入克隆位點組成完整LacO序列才會使菌落顯藍色,而基因閱讀框反向插入或空載體,菌落均呈白色。統計顯示,EGFP基因克隆至該載體并轉化后,藍色菌落占總菌落數的50%~60%,而且藍色菌落均為閱讀框正確插入的重組子(圖3)。
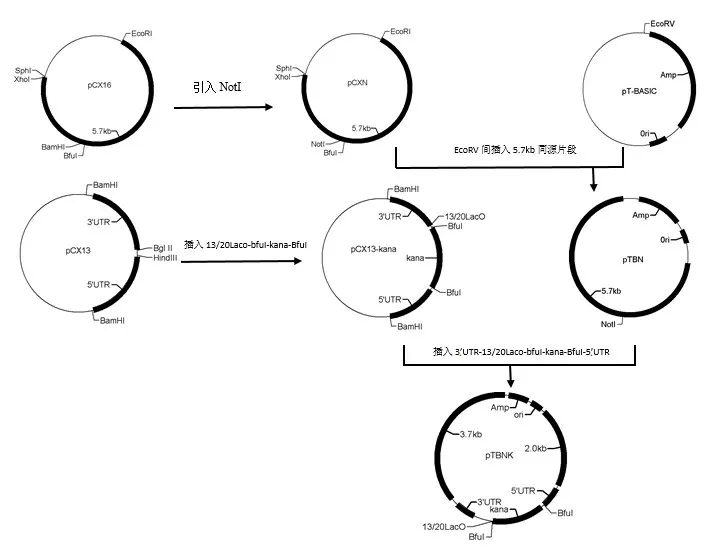
圖1 衣藻葉綠體定向表達T載體構建流程圖
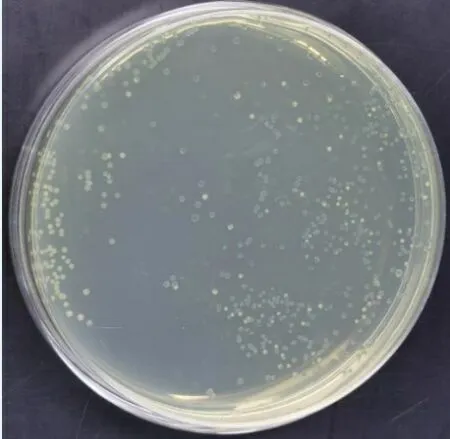
圖2 藍白斑篩選重組子平板

圖3 1:DNAmmaakkeerr 2Kpluuss IIII;2~2200:藍斑菌落PPCCRR擴增EEGGFFPP基因;2211~2255:白斑菌落PPCCRR擴增
本實驗構建衣藻葉綠體表達載體pTBNK,克隆效率高、篩選方法簡便、耗時短、成本低,而且不受酶切位點的影響,具備通用性,且重組子可通過藍白班篩選,優于傳統的兩步法酶切構建表達載體[7,11]。遺憾的是,本實驗中EGFP未表達。盡管如此,該構建方法極大簡化外源基因插入萊茵衣藻葉綠體表達載體的步驟,載體構建思路也可能適用于其他葉綠體表達載體的構建。
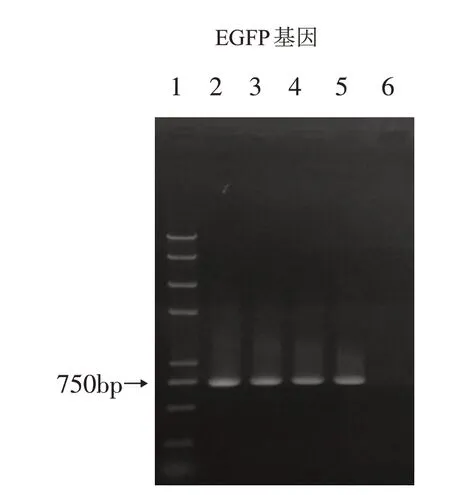
圖4 1:DNAmmaakkeerr 2Kpluuss IIII;2~5:轉基因衣藻抽提總DDNNAA作為模板PPCCRR 擴增EEGGFFPP 基因;6:野生型衣藻抽提總DDNNAA 作為模板PPCCRR擴增EEGGFFPP基因
