新型氮雜冠醚吸附劑對低濃度La3+的富集性能研究
2021-03-07 08:07:36吳江華肖松文楊天足張杜超萬洪強夏大廈
湖南有色金屬 2021年1期
吳江華,肖松文,楊天足,張杜超,萬洪強,夏大廈
(1.中南大學 冶金與環境學院,湖南 長沙 410083;2.長沙礦冶研究院有限責任公司,湖南 長沙 410012;3.杭州研趣信息技術有限公司,浙江 杭州 310012)
分子識別技術是一種基于超分子化學的新型高效選擇性分離技術,可針對目標離子的幾何尺寸、化學特性來選擇性設計合成配體分子,其中大環冠醚是研究最早、最透徹的一類陽離子識別主體化合物,已被廣泛應用于分離萃取、藥物合成、催化反應等領域[1]。美國IBC公司將冠醚負載于硅樹脂上合成SuperLig?-188樹脂,可從重稀土礦提取液中選擇性分離出純度為99.999%的Nd和Dy[2,3];截止到目前國內尚未出現采用分子識別技術富集分離稀土的相關報道。
稀土冠醚化合物的研究報道較多,但主要集中于硝酸稀土和氯化稀土體系,截至目前關于硫酸體系中稀土冠醚的研究尚未見報道[4,5]。前期研究發現,氮雜冠醚屬于中性含氮氧配體,在環上引入氮原子后可適當增大環腔內徑,能更好地通過腔穴尺寸匹配效應和氧氮聯合配位作用與稀土離子結合,對硫酸體系中輕稀土La3+表現出較強的選擇識別能力。由于冠醚具有一定的水溶性和化學毒性,采用常規的萃取分離不可避免會產生有機廢水的問題,同時弱化稀土離子的分離能力。試驗考慮將冠醚負載到大孔硅膠樹脂上,并深入研究固化冠醚對稀土離子的識別富集性能。
1 試 驗
1.1 試驗試劑
試驗所用試劑明細見表1。
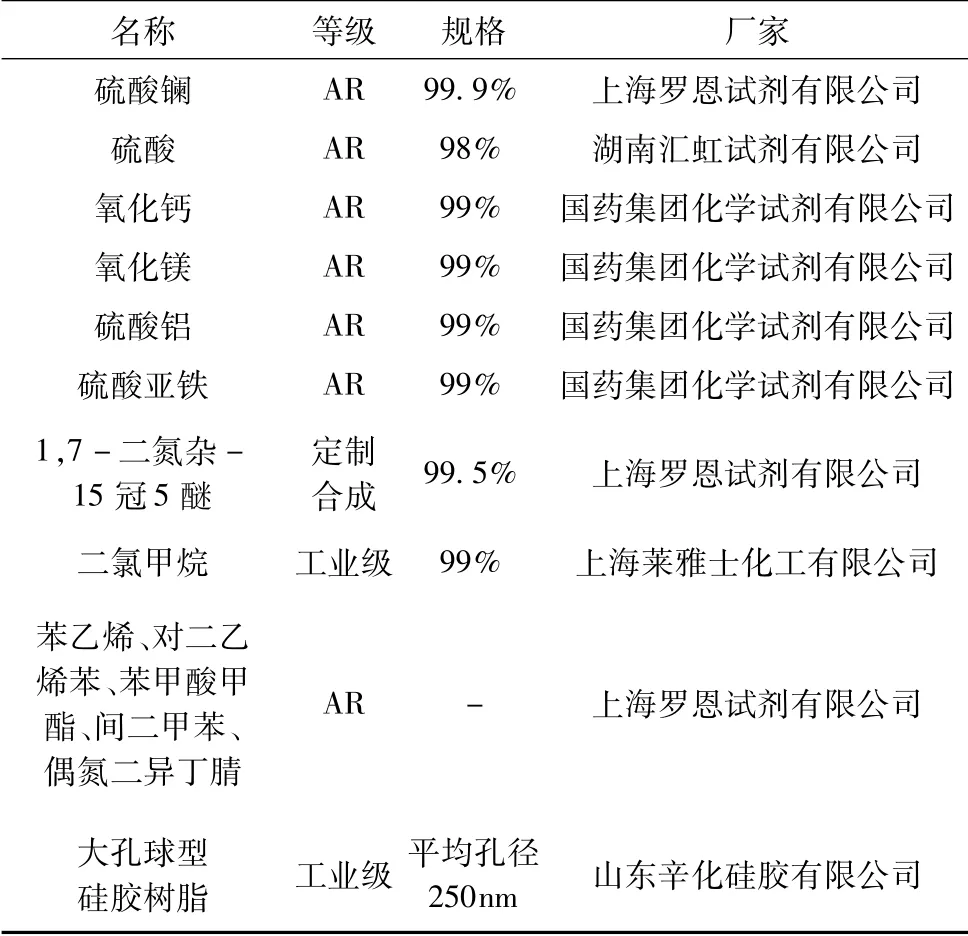
表1 試驗試劑明細
1.2 試驗儀器與設備
試驗所用儀器與設備的明細見表2。
1.3 試驗步驟
1.3.1 吸附劑的合成
1.硅膠樹脂的活化處理。100 g市售球型大孔硅膠樹脂,置于旋轉蒸發瓶中,通過橡膠導管加入適量85%苯乙烯、15%對二乙烯苯、苯甲酸甲酯(制孔劑)、間二甲苯(稀釋劑)、偶氮二異丁腈(引發劑),在N2保護、80℃恒溫下旋轉反應24 h。冷卻后取出過濾,樹脂采用丙酮和水洗滌3~5次,再加入200 mL甲醇完全浸泡24 h,重復處理兩次后將樹脂置于50℃真空干燥箱中干燥備用,即得活化硅膠樹脂。活化處理的目的是借助高分子有機物質的溶解侵蝕作用打開硅膠樹脂的面部孔隙,并將苯乙烯、對二乙烯苯等單體灌入樹脂內部孔隙中,起到打開內部孔隙的目的。甲醇一方面作為溶劑,可進一步去除進入硅膠顆粒內部的殘留雜質,同時可作為浸潤劑持續深入到硅膠樹脂結構內部,并借助表面張力增大樹脂內部孔隙,為后續吸附劑的合成提供更多的冠醚分子通道和化學接枝結合位點。

表2 主要儀器與設備
2.功能化冠醚吸附劑的合成。稱取50 g活化處理后的硅膠樹脂加入1 000 mL旋轉蒸發瓶中;再稱取2 g 1,7-二氮雜15冠5醚并溶解于200 mL二氯甲烷溶劑中,并全部轉移到旋轉蒸發瓶中。開啟旋轉蒸發儀,控制水溫為298 K,將轉速調至最大并旋轉反應30 min,使瓶內物料完全混合均勻;開啟旋轉蒸發儀的水冷和真空系統(設置真空度為0.1),重新開啟加熱并緩慢升溫到310 K,由于二氯甲烷的沸點為312.95 K,加熱過程中會觀察到液體沸騰狀態。待瓶中液體逐漸減少到有樹脂固體冒出,停止加熱,利用系統余溫將瓶內液體蒸干。該過程中一定要嚴格控制溫度和壓力,防止高真空下液體突然爆沸。負載冠醚后的硅膠樹脂顏色變深,將其取出,冷卻后用丙酮和純水清洗2~3遍,置于50℃真空干燥箱中干燥備用,即得功能化冠醚吸附劑。
1.3.2 樹脂靜態吸附試驗
按照不同固液比將功能化冠醚吸附劑與一定濃度的硫酸稀土溶液混合,于298 K下在恒溫振蕩箱中進行靜態吸附處理,取吸附后液用ICP-MS分析其中的La3+濃度,根據吸附處理前后溶液中金屬離子濃度變化來計算吸附劑對稀土離子的識別富集能力,并計算靜態吸附率K1和靜態飽和吸附容量Q1,計算公式為:
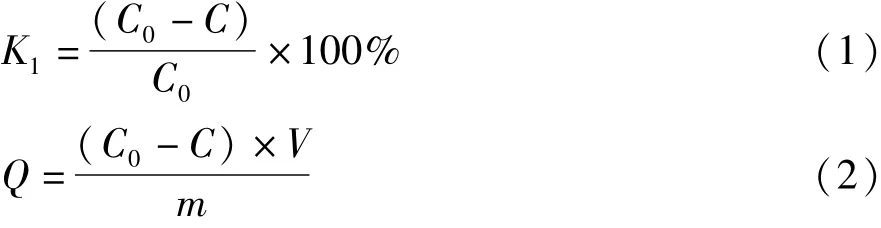
式中:C0、C分別為吸附前后液相中稀土離子的濃度/mg·L-1;V為液相體積/L;m為吸附劑質量/g。
2 結果與討論
2.1 新型吸附劑制備及吸附原理分析
與其它高分子載體相比,硅膠載體在耐酸堿腐蝕、機械性能等方面更具優勢。氮雜冠醚固化到硅膠樹脂的過程主要為物理作用,先借助活化劑擴展硅膠樹脂的內部孔隙,疏通大分子物質的進出通道,再采用真空吸入法,使大分子氮雜冠醚在溶劑的遷移作用下完全浸潤樹脂顆粒,并通過氫鍵形式與SiO2成鍵完成負載固化。基于物理作用對硅膠樹脂進行表面化學修飾,操作方法簡單,所制備的萃淋樹脂類吸附劑具有吸附容量高、結構穩定等優點,已成為低濃度有價金屬富集回收、重金屬污染治理、痕量金屬富集檢測的研究熱點[6]。
前期基于量子化學計算和試驗測定,已經確定氮雜冠醚可與La3+配位,形成La3+∶氮雜冠醚∶H2O=1∶1∶3的配合物,配位反應的機理在于冠醚環上的N、O原子、以及水分子上O原子與La3+之間的離子偶極相互作用,所形成的配合物結構如圖1所示,對應的萃取平衡常數K=105.46。氮雜冠醚固化到硅膠樹脂的過程不涉及改變冠醚結構的化學反應,因此能夠保持對La3+的選擇識別能力。
2.2 [La3+]的影響
為有效模擬南方離子吸附型稀土礦原地浸出尾液的溶液環境,配置一系列低濃度硫酸鑭溶液([La3+]≤0.6 g/L)。吸附劑用去離子水浸泡30 min后瀝干使用,設置吸附固(干吸附劑)液比為1 g/50 mL,控制吸附反應溫度25℃、時間30 min。考察了不同金屬離子初始濃度對其吸附率的影響,結果如圖2所示。
在固定吸附劑用量的情況下,只有當吸附量達到飽和吸附量水平后,靜態吸附率K1才隨著液相中La3+初始濃度的增大而減小。由此可計算得到,功能化冠醚吸附劑對的靜態飽和吸附容量Q1=22 mg/g。
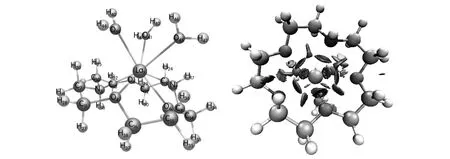
圖1 氮雜冠醚-La所形成配合物的結構圖
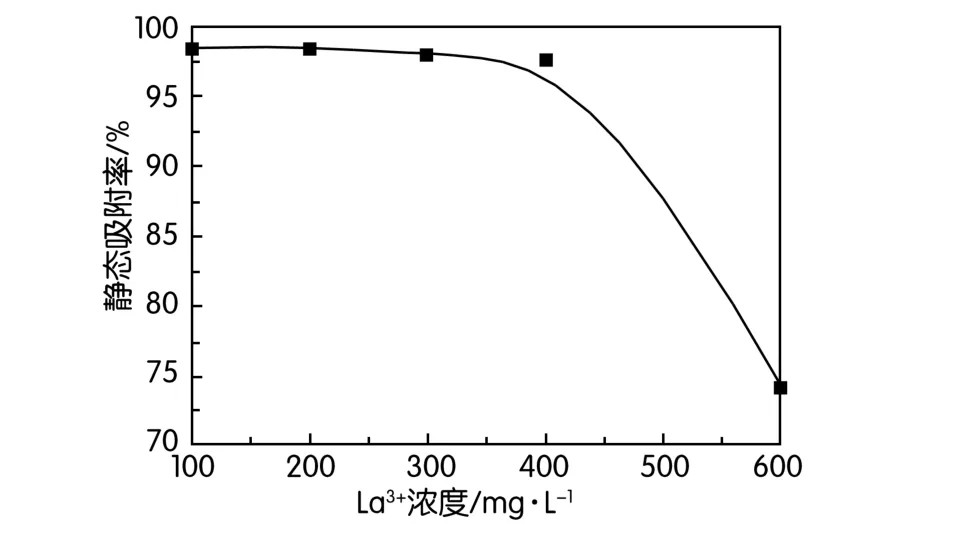
圖2 金屬離子濃度對靜態吸附率的影響
2.3 [SO]的影響
前期研究結果表明,在氮雜冠醚與La3+萃取過程中,溶液中的平衡陰離子會參與萃合物的組成。為驗證固化冠醚與它的配位結合是否受平衡陰離子的影響,固定液相[La3+]=0.3 g/L,固液比為1 g/50 mL,通過添加硫酸鈉來調節液相SO的濃度,并考察不同SO濃度對La3+吸附分離的影響,結果如圖3所示。

圖3 平衡陰離子濃度對靜態吸附率的影響
2.4 時間的影響
在[La3+]=0.3 g/L、[SO]=0.6 g/L、pH=2.0的溶液體系內,考察了常溫下吸附時間對La3+吸附分離的影響。對于大孔球型氮雜冠醚硅膠吸附劑而言,吸附反應過程可近似看作收縮未反應核模型,因此借助動力學方程對吸附過程進行了擬合分析,結果如圖4所示。
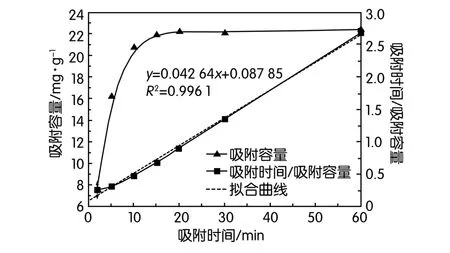
圖4 吸附動力學過程的數據擬合
由圖4數據可知,吸附反應速度很快,15 min左右即可達到吸附平衡。吸附劑與液相接觸的前10 min,吸附容量快速增長,主要原因在于吸附劑表面存在大量的可配位反應的結合位點,且溶液中La3+濃度大,離子遷移速度快,且吸附反應主要發生在吸附劑的外表面;隨著外表面結合位點趨于飽和,La3+需要進入吸附劑內部空腔,一方面吸附劑內部孔隙較小,且離子濃度變小,遷移速度變慢,另一方面已負載于吸附劑上的La3+會對溶液中游離La3+產生同電荷排斥作用,阻礙La3+向吸附劑內部遷移,從而表現為吸附速度變慢。
根據Lagergren方程對采用吸附過程進行擬合,結果發現吸附過程能很好地符合準二級吸附反應動力學方程,如式3所示:

式中:Qt、Qeq分別為t時刻與吸附達平衡時的吸附容量/mg·g-1。
相應的擬合直線方程為:y=0.042 6x+0.087 8(相關系數R2=0.996 1)。進一步計算可得平衡吸附容量Qeq=23.47 mg/g,對應的吸附反應速率常數k=0.020 7。根據準二級動力學方程的定義,可以推測氮雜冠醚吸附劑對La3+的吸附過程為化學吸附和物理擴散并存,且以化學吸附為主。
2.5 溫度的影響
為探討吸附熱力學過程,研究了298 K、318 K、338 K溫度條件下的吸附過程,液相體系中[La3+]=0.1~0.6 g/L、SO濃度為La3+的2倍、pH=2.0、固液比為2 g/100 mL,吸附時間為15 min。并借助吸附等溫方程對試驗結果進行擬合分析,結果如圖5所示。
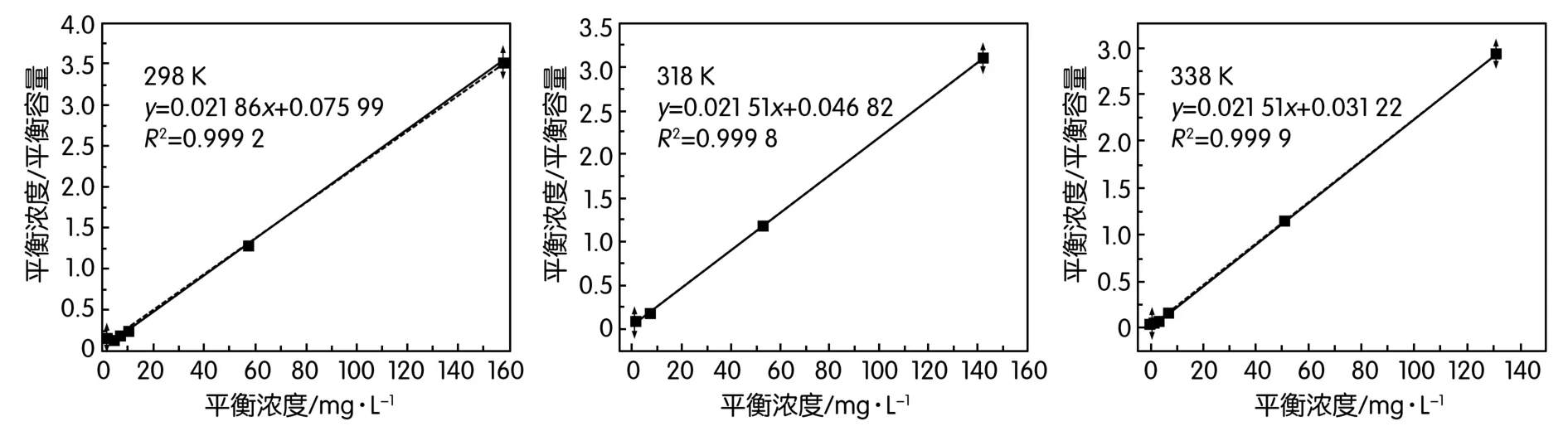
圖5 不同吸附溫度下的等溫吸附數據擬合
吸附過程能夠很好地符合Langmuir等溫吸附方程,不同溫度下的吸附等溫擬合方程分別如下:
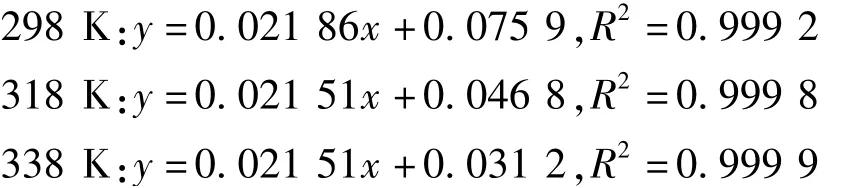
結合Langmuir方程的定義,可計算出298 K、318 K、338 K下的最大飽和吸附容量分別為22.62 mg/g、22.94 mg/g、23.26 mg/g,說明吸附反應屬于吸熱過程。從吸附容量變化來看,溫度對吸附率的影響較小,因此從過程節能的角度來考慮,298 K較佳。
2.6 吸附機理探討
為進一步探究氮雜冠醚吸附劑對La3+的吸附機理,采用紅外光譜對吸附反應前后的吸附劑進行表征,所得FTIR譜圖如圖6所示。
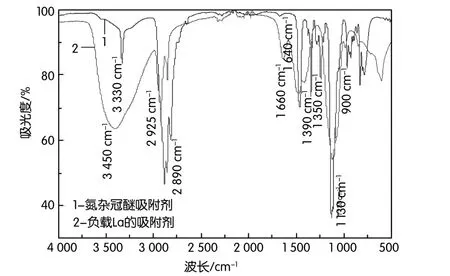
圖6 負載La3+前后的氮雜冠醚吸附劑的紅外光譜圖
負載La3+前后的吸附劑,在紅外光譜上的最大差異體現在兩點:(1)位于3 330 cm-1、1 640 cm-1、900 cm-1處的N-H特征峰,在負載La3+以后明顯變弱;(2)1 250 cm-1處的C-N特征峰,在配位反應之后出現藍移,并與1 120 cm-1處的-C-O-C-特征峰疊加,峰強和峰寬明顯增強。結合氮雜冠醚本身結構來分析,負載La3+以后有機物的結構發生變化,說明N、O原子均參與了配位反應,且由于N、O均屬于給電子配體,基于誘導效應致使相鄰-CH2基團的特征峰向高波數遷移,體現在吸附劑的2 890 cm-1、1 350 cm-1處的特征峰,在負載有機物中分別遷移到2 925 cm-1、1 390 cm-1處。
3 結 論
1.將功能化冠醚固化到硅膠樹脂上所制成的新型吸附劑,對硫酸體系中稀土La3+表現出較強的吸附分離能力。
2.最佳吸附條件為溫度298 K、時間15 min,且液相中平衡陰離子濃度和溶液pH對吸附過程影響不大。吸附反應為吸熱過程,以化學吸附為主,最大飽和吸附容量為22~23 mg/g。
3.吸附反應的機理在于冠醚環上N、O原子與La3+之間的配位反應。
