Fe3O4@MOF-808磁性固相萃取結合高效液相色譜法測定大米中3種二苯醚類除草劑
2021-03-10 09:21:48賈葉青齊沛茹王雅慧冀欠欠王曼曼郝玉蘭
色譜 2021年3期
閆 萌, 賈葉青, 齊沛茹, 王雅慧, 冀欠欠, 王曼曼, 王 茜, 郝玉蘭
(華北理工大學公共衛生學院, 河北 唐山 063210)
二苯醚類除草劑(diphenyl ether herbicides, Des)是一種原卟啉原氧化酶抑制劑,主要用于防除一年生和多年生闊葉雜草,是水稻田主要的除草劑品種之一[1]。Des在環境中具有累積性和持久性,對生物體的影響不容忽視。研究表明,大部分Des會對眼睛和皮膚產生刺激作用[2]。除草醚可以引起高鐵血紅蛋白血癥、溶血性貧血和黃疸等疾病;甲羧除草醚對魚及水生動物具有高毒作用。常見的Des有除草醚(nitrofen, NIT)、乙氧氟草醚(oxyfluorfen, OXY)和甲羧除草醚(bifenox, BIF),其結構式見圖1。國標GB 2763-2019[3]對水稻種植過程中Des的使用及大米中Des的殘留限值做出了規定,規定糙米中OXY的最大殘留限量為0.05 mg/kg。因此,及時掌握大米中的Des殘留情況,建立快速、可靠的大米中Des檢測方法具有重要意義。
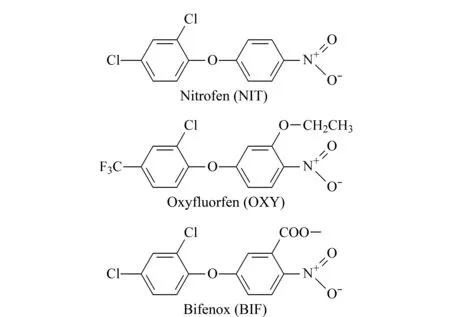
圖 1 3種二苯醚類除草劑的化學結構式Fig. 1 Chemical structures of the three diphenyl ether herbicides (Des)
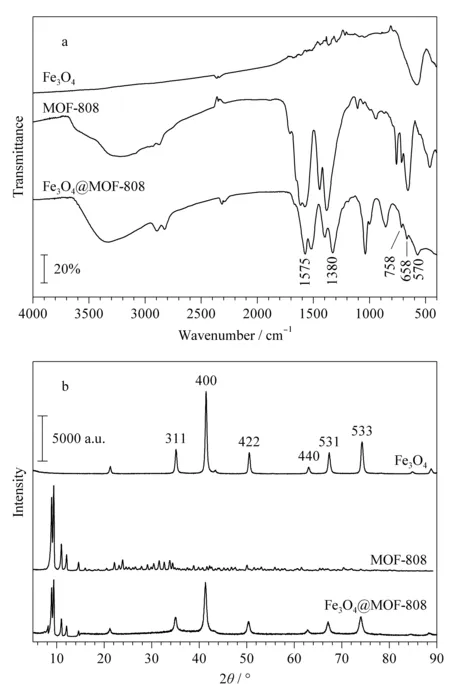
圖 2 Fe3O4、MOF-808和Fe3O4@MOF-808的(a)紅外光譜圖和(b)X射線衍射圖Fig. 2 (a) FT-IR spectra and (b) X-ray diffraction (XRD) patterns of Fe3O4, MOF-808 and Fe3O4@MOF-808
目前,常采用高效液相色譜法(HPLC)、氣相色譜法和氣相色譜-質譜法等分析谷物中的Des[4-6]。由于大米樣品基質復雜,且目標分析物在樣品中的含量較低,不易檢出。在儀器分析前,需要對樣品進行充分凈化以去除雜質干擾,同時對目標物進行富集。大米樣品中除草劑殘留的前處理方法主要有固相萃取和分散固相萃取[4,7]。磁性固相萃取(magnetic solid phase extraction, MSPE)屬于分散固相萃取技術中的一種,其通過施加外部磁場可以實現吸附劑與樣品溶液的快速分離[8]。與傳統填充柱相比,MSPE具有操作步驟簡便、基質干擾小、無需填充萃取柱以及避免柱壓力等優點[9]。MSPE吸附劑是由磁性核心和具備吸附作用的吸附材料組成,其中磁性核心一般采用Fe3O4,具備吸附作用的有機基團可以根據待測物進行靈活選擇。吸附材料是決定磁性吸附效果的關鍵。
金屬-有機骨架(metal organic frameworks, MOFs)是以有機配體中的氧原子和氮原子連接無機金屬離子形成的多孔晶體材料,具有比表面積大、溶劑穩定性好以及孔道可調控等特性而備受關注[10]。以Zr金屬為中心的MOFs具有良好的熱穩定性與化學穩定性,是近年來發展迅速的一種新型吸附劑。MOF-808由氯化鋯和1,3,5-苯三甲酸制備而成,以Zr6O4(OH)4(-CO2)6(HCOO)6為二級構建單元,與6個1,3,5-苯三甲酸單元連接,通過橋連作用形成超四面體結構[11]。該四面體籠的內孔尺寸為0.48 nm,由10個連續的四面體籠形成大金剛烷籠的內部孔徑為1.84 nm,比表面積可以達到2 060 m2/g[12]。同時,MOF-808結構中豐富的金屬位點及有機配體可以提供π-π共軛、氫鍵作用,使其具備良好的吸附分離功能[13,14]。
本研究利用溶劑熱法將磁性核心Fe3O4與MOF-808進行組裝,構筑Fe3O4@MOF-808吸附劑,可同時發揮MSPE技術優勢及MOF-808的優良性能。將其用于大米中3種Des的前處理,結合HPLC-UV,建立了簡單、快速測定大米中3種Des的分析方法。
1 實驗部分
1.1 儀器和試劑
Agilent 1200型高效液相色譜-紫外檢測器(美國Agilent公司); JEM-2800F聚焦離子束-電子束雙束電鏡(美國FEI儀器有限公司); FTIR-8400S傅里葉變換紅外光譜儀(日本Shimadzu公司); D8-Venture單晶X射線衍射儀(德國布魯克公司); 9600-1振動樣品磁強計(美國LDJ公司); 100 mL-水熱合成反應釜(天津凱易達儀器設備銷售有限公司); ET-3301A型氮氣濃縮裝置(上海歐陸科儀有限公司)。
1,3,5-苯三甲酸(H3BTC)、氯化鋯(ZrCl4)、N,N′-二甲基甲酰胺(DMF)、甲酸和乙二醇(純度99%,上海阿拉丁試劑有限公司);六水合氯化鐵(FeCl3·6H2O)、三水合乙酸鈉(NaOAc·3H2O)和無水乙醇(純度99%,天津光復科技有限公司);甲醇(methanol, MeOH)和乙腈(acetonitrile, ACN)(色譜純,美國Thermo Fisher Scientific公司);娃哈哈純凈水(杭州娃哈哈集團有限公司)。
標準物質NIT(純度99.0%)和OXY(純度99.5%)(上海阿拉丁試劑有限公司); MeOH配制的100 μg/mL BIF(純度99.0%,北京百靈威科技有限公司)。
大米樣品購自唐山市當地超市。
1.2 標準溶液的配制
分別稱取NIT和OXY 1 mg,使用MeOH將其配制成100 μg/mL的混合標準儲備液,將該儲備液與BIF標準溶液(100 μg/mL)按照1∶1的體積比配制為50 μg/mL的NIT、OXY和BIF混合標準溶液,4 ℃避光保存。
1.3 Fe3O4@MOF-808吸附劑的構筑
通過溶劑熱法制備MOF-808[15]。稱取174.8 mg ZrCl4(0.75 mmol)和52.5 mg H3BTC(0.25 mmol)置于燒杯中,依次加入8 mL DMF和8 mL甲酸,超聲20 min,將混合溶液移入反應釜中,置于預先加熱至120 ℃的烘箱中,反應24 h。反應完畢后于烘箱中自然冷卻至室溫,離心收集沉淀,采用DMF(30 mL×2)、丙酮(30 mL×2)和MeOH(30 mL×3)依次洗滌,將所得白色粉末于100 ℃真空干燥。
將93.2 mg MOF-808分散于20 mL乙二醇中,超聲3 h;取270.0 mg FeCl3·6H2O和700.0 mg NaOAc·3H2O溶解于另一20 mL乙二醇中,超聲3 h;將兩種溶液混合,超聲1 h;將混合溶液移入反應釜,置于預先加熱至200 ℃的烘箱中,反應24 h。反應完畢后于烘箱中自然冷卻至室溫,利用外加磁場分離產物,并用適量乙醇和去離子水清洗產物,直至上清液清澈透明,最后60 ℃真空干燥12 h,得到Fe3O4@MOF-808吸附劑。
1.4 樣品制備
準確稱取1 g大米樣品,充分粉碎,過篩,轉移至50 mL離心管中,加入2.5 mL MeOH,超聲10 min進行提取,以10 000 r/min離心5 min,上清液轉移至樣品瓶中,超純水定容至15 mL,待用。
1.5 磁性固相萃取流程
稱取25 mg Fe3O4@MOF-808吸附劑,置于處理好的15 mL大米樣品溶液中,混勻器以1 000 r/min混勻吸附6 min,施加外部磁場將吸附劑與樣品溶液分離,棄去上清液。采用0.5 mL MeOH進行洗脫,充分振蕩2 min后磁分離,重復2次,合并洗脫溶液,室溫下氮吹濃縮至干,MeOH定容至0.5 mL,待測。
1.6 色譜分離條件
色譜柱為Agilent XDB C18柱(150 mm×4.6 mm, 5 μm);柱溫為35 ℃;流動相為(A)H2O和(B)MeOH;流速為1 mL/min。洗脫程序為0~5 min, 88%B~89%B,運行時間5 min。進樣量為10 μL;檢測波長為208 nm。
2 結果與討論
2.1 Fe3O4@MOF-808吸附劑的表征
通過FT-IR對制備的Fe3O4、MOF-808和Fe3O4@MOF-808復合材料的基團構成進行表征。如圖2a所示,Fe3O4@MOF-808復合材料在658 cm-1和758 cm-1處的吸收峰對應MOF-808中Zr-O的外彎曲振動,1 380 cm-1處的吸收峰歸于MOF-808中Zr-O-H基團,1 575 cm-1處的吸收峰為MOF-808中-COOH的彎曲振動所致[16]; 570 cm-1處的吸收峰歸因于Fe3O4中Fe-O-Fe的伸縮振動[17]。通過XRD對制備的Fe3O4、MOF-808和Fe3O4@MOF-808復合材料的晶體結構進行了表征。如圖2b所示,Fe3O4@MOF-808在35.54°、41.12°、50.75°、63.22°、67.89°和74.15°處出現了特征峰,與Fe3O4相比,特征峰的位置相同,對應Fe3O4的(311)晶面、(400)晶面、(422)晶面、(440)晶面、(531)晶面和(533)晶面;同時在8.32°和8.69°處出現了與MOF-808譜圖對應的特征峰,與文獻[18]報道一致。

圖 3 (a)MOF-808和(b)Fe3O4@MOF-808的SEM圖Fig. 3 SEM images of (a) MOF-808 and (b) Fe3O4@MOF-808
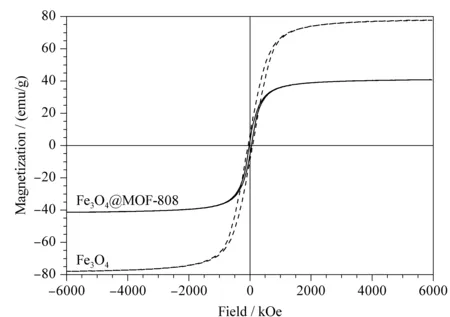
圖 4 Fe3O4和Fe3O4@MOF-808的磁滯曲線圖Fig. 4 Magnetization curves of Fe3O4 and Fe3O4@MOF-808
對制備的Fe3O4@MOF-808進行掃描電鏡分析,觀察其形貌。如圖3a所示,MOF-808具有規則的八面體形貌,粒徑范圍在400~500 nm之間,表面較光滑且分布均勻;球形的Fe3O4納米顆粒分散于MOF-808表面(見圖3b)。采用振動樣品磁強計對制備的Fe3O4和Fe3O4@MOF-808的磁性進行表征。結果如圖4所示,Fe3O4@MOF-808沒有明顯的磁滯現象,其飽和磁化強度為40.35 emu/g,雖然較Fe3O4的飽和磁化強度78.26 emu/g有所下降,但仍然可以滿足快速磁分離的要求。
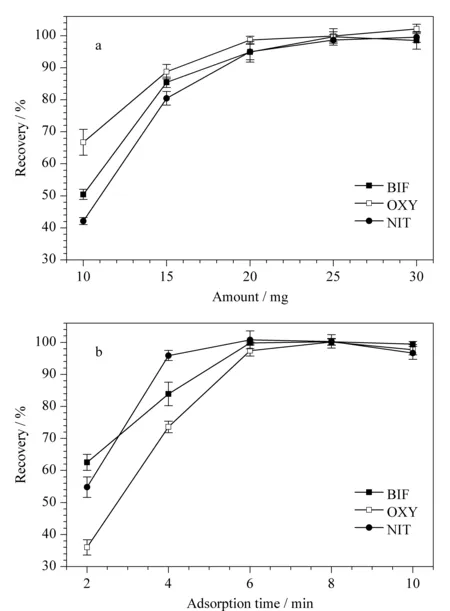
圖 5 (a)吸附劑用量和(b)吸附時間對Des回收率的影響(n=3)Fig. 5 Effects of (a) amount of sorbent and (b) adsorption time on the recoveries of Des (n=3)
2.2 磁性固相萃取條件的優化
吸附和洗脫過程是影響MSPE效果的關鍵,為了獲得最優的萃取條件,實驗對吸附劑用量、吸附時間、洗脫溶劑種類和洗脫體積進行了優化。采用15 mL MeOH-水(1∶5, v/v)混合標準溶液進行實驗,3種Des濃度均為65 ng/mL,所有實驗平行測定3次。
2.2.1吸附劑用量和吸附時間
實驗分別考察了吸附劑用量為10、15、20、25和30 mg,吸附時間為2、4、6、8和10 min時,3種Des回收率的變化情況。結果如圖5a所示,當吸附劑用量為25 mg時,回收率最高。繼續增加吸附劑用量,回收率無明顯變化。當吸附時間為6 min時,3種Des的回收率均達到95%以上,表明吸附已經達到飽和。因此,實驗選擇25 mg為最優吸附劑用量,6 min為最優吸附時間。
2.2.2洗脫溶劑和洗脫體積
根據“相似相溶”原理,本實驗選取MeOH、ACN和丙酮作為洗脫溶劑。實驗固定吸附劑用量為25 mg,吸附時間為6 min,洗脫體積0.5 mL×2,對3種洗脫溶劑進行考察。結果如圖6a所示,當洗脫溶劑為MeOH時,回收率最接近100%。實驗同時考察了不同洗脫體積(0.5 mL、0.5 mL×2、0.5 mL×3和0.5 mL×4)對Des回收率的影響,結果如圖6b所示,當洗脫體積為0.5 mL×2時,3種Des的回收率達到最佳。因此選用0.5 mL×2的MeOH作為最佳洗脫溶劑。
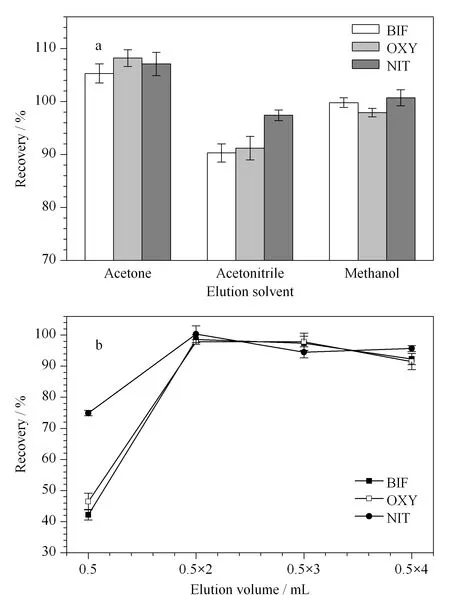
圖 6 (a)洗脫溶劑種類和(b)洗脫體積對3種Des 回收率的影響(n=3)Fig. 6 Effects of (a) elution solvent and (b) elution volume on the recoveries of Des (n=3)
2.3 方法驗證
2.3.1線性范圍、檢出限和定量限
在優化的實驗條件下,按照1.4節方法處理空白大米樣品,得到空白基質溶液,向其中添加一定量的混合標準溶液配制成質量濃度為2、4、20、40、80、200和300 μg/L的系列基質混合標準溶液,以峰面積(y)對除草劑的質量濃度(x, μg/L)繪制標準曲線。結果如表1所示,NIT、OXY和BIF在2~300 μg/L范圍內線性關系良好,相關系數均大于0.998。以信噪比(S/N)=3確定3種除草劑的檢出限(LOD),以(S/N)=10確定3種目標物的定量限(LOQ),結果見表1。
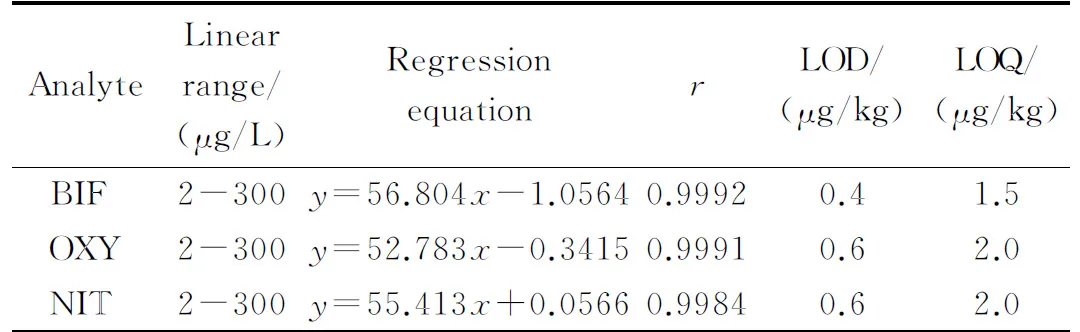
表 1 3種Des的線性范圍、檢出限和定量限
為了驗證方法的準確度和精密度,對空白大米樣品進行加標回收試驗,3種Des的加標水平分別為5、10和20 μg/kg,每個加標水平平行測定3次。由表2可知,本方法對3種Des的回收率為87.3%~96.7%,日內和日間精密度(n=3)分別為2.9%~8.2%和4.2%~10.8%,表明本方法的準確度和精密度良好,能夠滿足分析要求。
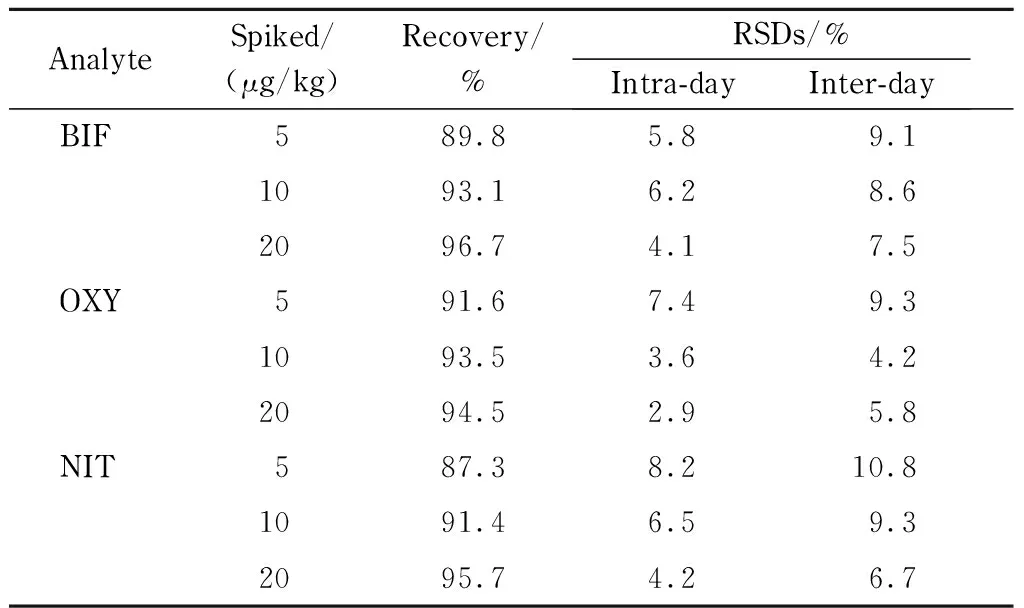
表 2 本方法的回收率和精密度(n=3)
2.4 實際樣品分析
采用最佳的實驗條件(25 mg吸附劑吸附6 min),吸附劑與目標物可達到吸附平衡,選擇0.5 mL×2的MeOH作為洗脫溶劑,可以將目標物從吸附劑上完全洗脫,結合高效液相色譜法,測定10個大米樣品中3種Des,測定結果見表3。所有樣品中均未檢出這3種Des。通過對每種樣品進行加標回收試驗證實方法的準確性,結果顯示,實際樣品中3種分析物的加標回收率(加標水平為10 μg/kg)為85.7%~95.4%, RSD≤11.3%,滿足實際樣品的分析要求。
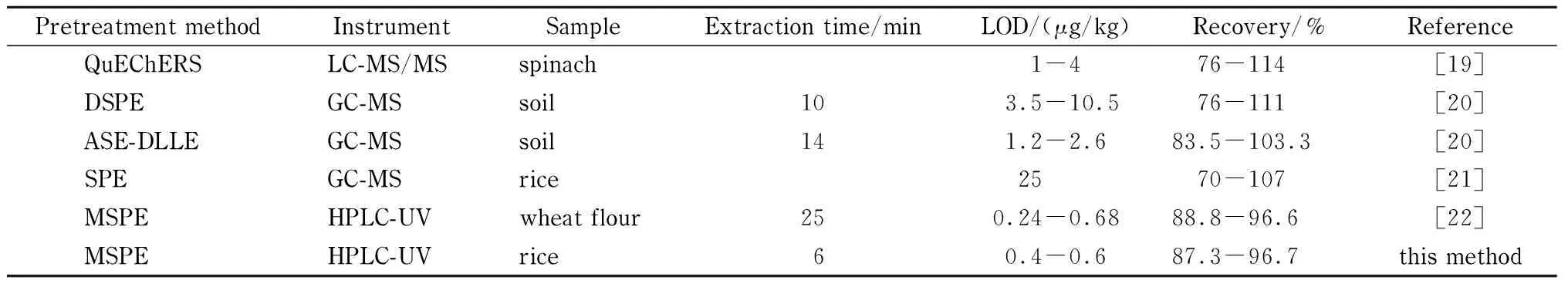
表 4 本方法與國家標準方法及文獻方法對Des分析結果的對比

表 3 大米樣品中3種Des的分析結果及加標回收率
在最優條件下,將3種Des按照20 μg/kg水平加入大米樣品,結合HPLC-UV進行分析。圖7b為經過粉碎過篩,加入MeOH提取處理的大米樣品直接進樣的分析結果,雜質峰明顯,3種Des均未檢出;而經過本方法處理后,結果如圖7c所示,雜質峰降低,在對應出峰位置均檢出樣品峰,說明本方法能夠有效富集凈化大米樣品中的3種Des。在最優條件下,通過對比最終定容的甲醇溶液中3種Des的濃度與大米樣品溶液中原始目標物濃度之比,計算本方法對3種Des的富集因子,結果在25~29之間。
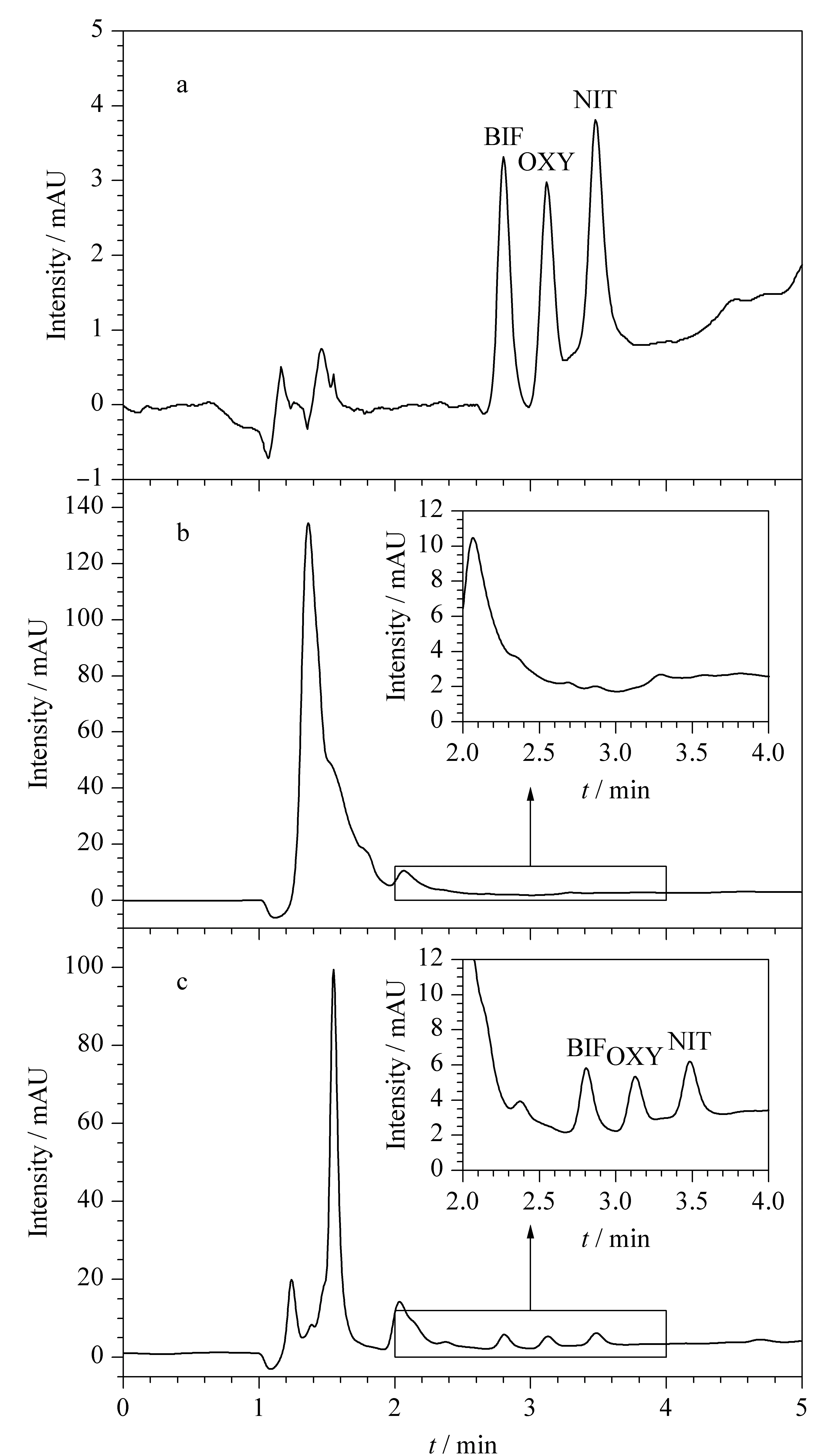
圖 7 (a)Des標準溶液以及加標大米樣品(20 μg/kg)(b)直接進樣和(c)經本方法前處理后進樣的色譜圖Fig. 7 Chromatograms of (a) Des standard solution, spiked rice samples (20 μg/kg) (b) direct injection and (c) pretreatment with the proposed MSPE method
2.5 與文獻方法對比
將本方法與國家標準方法以及相關文獻報道方法進行對比(見表4)。結果顯示,本方法的檢出限為0.4~0.6 μg/kg,優于國家標準方法,且與其他分析方法相比檢出限和回收率相當。在操作時長和簡便性方面,本方法使用25 mg吸附劑僅需6 min即可完成對大米樣品中Des的吸附,MSPE無需額外的離心、淋洗等步驟,操作簡單。
3 結論
本研究采用溶劑熱法成功制備了Fe3O4@MOF-808磁性復合材料,將其用作固相萃取吸附劑,結合HPLC-UV分析了大米樣品中的3種Des。該方法操作簡單、快速,滿足大米樣品中3種Des的分析要求,為大米樣品中除草劑的殘留分析提供了新的選擇。
猜你喜歡
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
今日農業(2019年15期)2019-01-03 12:11:33
電子制作(2018年18期)2018-11-14 01:48:24
現代園藝(2017年19期)2018-01-19 02:50:21
山東工業技術(2016年15期)2016-12-01 05:31:22
意林原創版(2016年10期)2016-11-25 10:28:30
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
營銷界(2015年23期)2015-02-28 22:06:18
