CCSD(T)study on the structures and chemical bonds of AnO molecules(An=Bk-Lr)?
2021-03-19 03:21:58XiyuanSun孫希媛PengfeiYin殷鵬飛KaimingWang王開明andGangJiang蔣剛
Chinese Physics B 2021年3期
Xiyuan Sun(孫希媛), Pengfei Yin(殷鵬飛), Kaiming Wang(王開明), and Gang Jiang(蔣剛)
1College of Science,Sichuan Agricultural University,Ya’an 625014,China
2Institute of Atomic and Molecular Physics,Sichuan University,Chengdu 610065,China
Keywords: ab initio calculation,AnO(Bk-Lr)molecules,density functional theory(DFT),chemical bonds
1. Introduction
Actinides (An) are very heavy elements in the periodic table. Among the actinide elements, Th, U,and the transuranium elements of Np, Pu, Am and Cm have important applications and have been synthesized in appreciable quantities in nuclear reactors.[1-3]The other actinides are mostly used for research. Most of them are radioactive. Therefore,the experimental studies are not easy. Computational modeling is particularly useful for systems. Quantum chemistry can model molecular properties of species containing actinides, leading to an improved understanding. However, the studies for the latter actinides (Bk-Lr) are very rare because these elements are not present in nature. The energy levels and atomic spectra of neutral and ionized Bk, Cf, and Es have been determined in the spectroscopic study.[4]Moreover,the first ionization energies of Bk-Es were studied by resonance ionization mass spectroscopy(RIMS).[5]
Theoretical studies on the latter actinide compounds are also rare. The first and second ionization potentials of Bk-Lr atoms were obtained with multireference averaged coupled-pair functional (ACPF) calculations in conjunction with extended valence basis sets.[6]The bond lengths, binding energies, and vibrational constants of the monohydride,monoxide, and monofluoride of lawrencium (Lr) were determined at ab initio level of theory.[7]The bond distances and dissociation enthalpies of actinide oxides AnO and AnO2(An=Th-Lr) were predicted by density functional theory(DFT) methods in combination with the relativistic smallcore pseudopotentials.[8]It is found that the high-level ab initio calculations were rare for the latter actinide oxides AnO(An=Th-Lr).
In the present work, we have performed the ab initio calculations,second-order Moller-Plesset perturbation theory(MP2) and coupled-cluster single-, double-, and perturbative triple-excitations[CCSD(T)],on the molecular structures,dissociation energies, bonding natures of AnO (An=Bk-Lr)molecules. Different DFT functionals were also employed for these species, and we would like to pick out the more appropriate functionals for the present species by comparing with CCSD(T)results.
2. Calculational details
The molecular geometries of AnO (Bk-Lr) molecules were optimized using the ab initio methods, MP2 and CCSD(T). The Cologne-Stuttgart small-core scalar relativistic effective core potential(SC-RECP)in combination with the associated basis set[6,7]was used to describe the heavy atoms(Bk-Lr), and the triple-ζ valence basis set of def2-TZVP[9]was used for the O atoms. The ab initio calculations were performed with the ORCA program.[10]Four hybrid functionals,including B3LYP,[11]M06-2X,[12]PBE0,[13]and TPSSh,[14]were also employed for comparison. We selected these four functionals because they were successful used in the studies related to the actinide complexes in previous works.[15,16]Furthermore, Hartree-Fock (HF) exchange proportions in these functionals are different, and they can be used to evaluate the influences from HF exchange proportion on the calculational results.The DFT calculations were performed with G09 package,[17]in which the correlation-consistent cc-pVTZ basis set was applied for O atoms. The electronic states with different spin multiplicities were calculated for each system to determine the ground electronic state. In the CCSD(T) calculations, the DFT starting orbitals were used. The bonding nature of An-O chemical bonds was revealed by the quantum theory of atoms in molecules(QTAIM),[18]as implemented in the MULTIWFN program.[19]Natural bonding orbital(NBO)analyses[20]were performed with NBO version 3.1 incorporated in G09 package to obtain the charges, bond orders and orbital populations of titled molecules.
3. Results and discussion
The geometry optimizations of the titled AnO molecules were performed with MP2, CCSD(T), and four hybridization functionals (B3LYP, M06-2X, PBE0, and TPSSh), respectively. The predicted bond lengths, vibrational frequencies,and dissociation energies(D0)of AnO(An=Bk-Lr)series were collected in Table 1. The dissociation energy D0defined as the difference between AnO molecules and free An and O atoms were calculated with the equation D0=E(An)+E(O)?E(AnO), where E(An), E(O),and E(AnO)are the energies of free An and O atoms, and AnO molecule,respectively.

Table 1. The bonding lengths Re, vibrational frequencies ωe, and dissociation energies D0, and spin multiplicity (M) of AnO (An=Bk-Lr)molecules predicted with different calculational methods.



Table 2 shows the natural population analysis (NPA)charges of An atoms,Wiberg bond indexes(WBI),and population in different shells of AnO (An=Bk-Lr) molecules obtained at M06-2X level of theory. One can find that the An atoms in the titled species transfer more than 1|e| to the O atom, and act as the electron donor. The significant electron transfers from An to O atoms imply the ionic character of An-O bonds. The single bond characters of AnO bonds are revealed by the WBI values. The populations in different shells of An atoms in the titled species are collected in Table 2. It can be found that the 7s orbitals in the studied molecules are single-electron occupation. Moreover, it is important to note that the 6d orbitals are also partially occupied. Thus, the 7s,6d, and 5f atomic orbitals participate in chemical bonding of AnO(Bk-Lr)together.
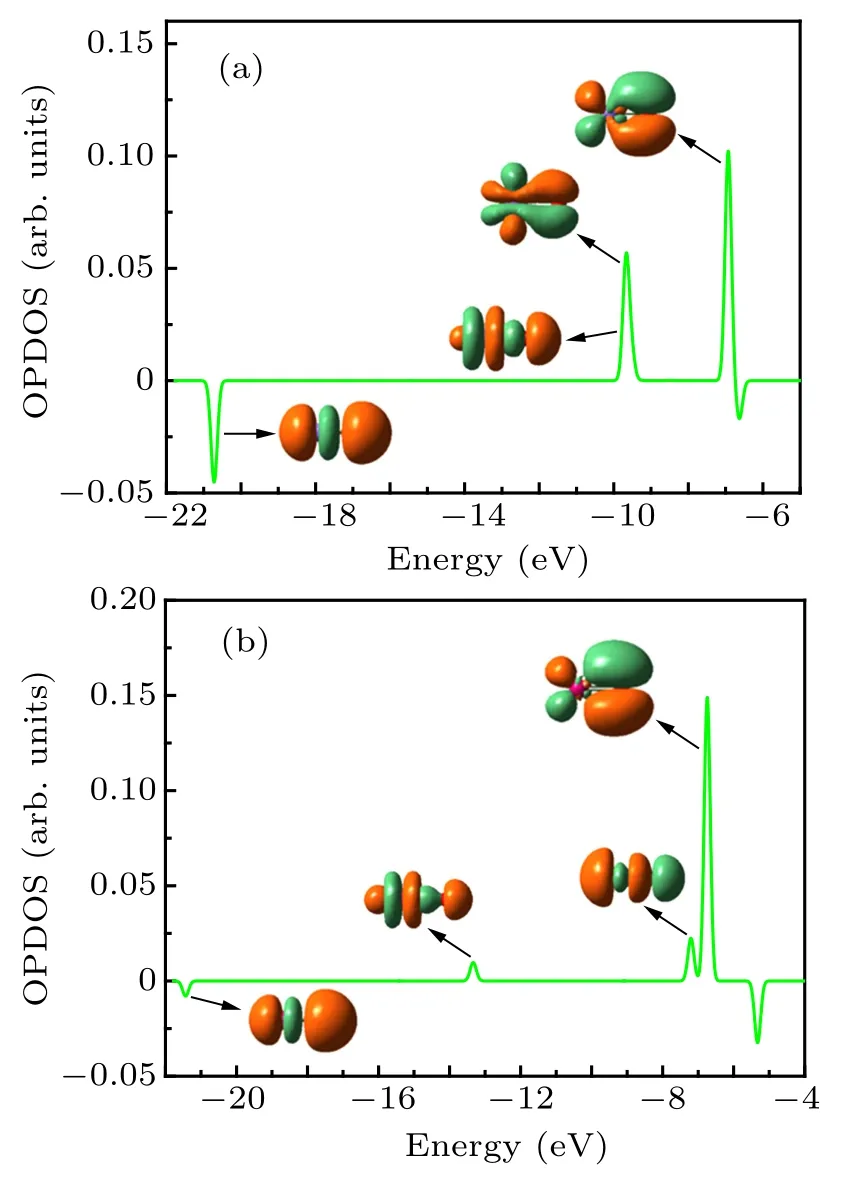
Fig.1. The overlap population density of states(OPDOS)and molecular orbitals relevant to An-O bonds of BkO(a)and LrO(b)molecules.
The overlap population density of states (OPDOS)analyses[21-23]were performed to reveal the bonding nature of AnO (Bk-Lr) molecules. For BkO-NoO molecules, the bonding orbitals are similar (see Figs. S1-S5 in the Supplementary Material). Therefore,we only depict the OPDOS and relevant molecular orbitals in Fig.1 for BkO and LrO,respectively. As shown in Fig.1(a),two positive OPDOS peaks are localized around ?10 eV and ?7 eV, respectively. The peak at high energy level is identified as π bonds from the overlaps of Bk 6d and O 2p atomic orbitals. The 5f atomic orbitals of Bk are overlapped with O 2p, resulting in the π bond and σ bond at ?10 eV.In addition to the bonding orbitals, the antibonding orbitals from Bk 6p and O 2s are also formed in the low-energy region. Previous work[22,23]also found this type of anti-bonding orbital in other actinide complexes. As for the LrO molecule, the predominant positive OPDOS peak shows that a π bond nature is mainly derived from the Lr 6d and O 2p atomic orbitals. The overlaps of Lr 7s and O 2p result in the σ bond near ?7 eV.The weak OPDOS peak corresponding to the σ bond from Lr 5d and O 2p is found around ?13 eV.The antibonding orbital is also formed.
The chemical bonding characters of AnO molecules were analyzed within the quantum theory of atoms in molecules(QTAIM)[18]framework. The wavefunction used in the QTAIM analyses was obtained at M06-2X level, which predicted the same ground electronic state as the MP2 method.The topological parameters relevant to (3, ?1) bond critical points(BCP)of An-O bonds are shown in Table 3,and the 2D plots of electron density are depicted in Fig.2. The topological parameters shown in Table 3 include the electron density at BCP (ρ(r)), Laplacian of electron density (?2ρ), the total electron energy density H(r) (defined as the sum of local kinetic energy density G(r)and the local potential energy density V(r)). Other important ratios (H(r)/ρ(r),?V(r)/G(r)),are used to assess the covalency. According to the criteria of QTAIM,the An-O bonds exhibit typical closed-shell characteristic, which can be concluded from the positive Laplacian?2ρ(r) value. Meanwhile, we also note that the partial covalent interaction of An-O bonds is revealed by the negative H(r)value and ?V(r)/G(r)ratio larger than 1. Energy density per electron, H(r)/ρ(r), which is called “bond degree”parameter(BD).[24]The BD can render the covalence degree.The stronger the interaction, the greater the BD magnitude.From BkO to NoO, the absolute values of BD are gradually decreased, suggesting the weakened bonding strength. One can see from Fig.2 that the electron density at BCP is also gradually decreased from BkO to NoO.The electron localization function(ELF)[25]and localized orbital locator(LOL)[26]at the BCP are also shown in Table 3. We can see that the ELF and LOL correlate excellently with electron density and total electron energy density H(r). On the whole,the An-O bonds favor an ionic character in nature, and they also show partial covalent nature.

Table 3. The topological parameters of AnO(An=Bk-Lr)molecules.

Fig.2. The 2D plots for the electron density of AnO (An=Bk-Lr)molecules.
4. Conclusion
In summary, we have obtained the molecular geometries and dissociation energies of AnO (Bk-Lr) molecules at the CCSD(T)level of theory with MP2 starting orbitals. The DFT calculations have also been performed for these molecules with four HF/DFT hybridization functionals, B3LYP, M06-2X,TPSSh, and PBE0, for the sake of comparison. By comparing with CCSD(T) results, we conclude that the M06-2X functional generally gives the lower dissociation energies, on the other hand, the MP2 highly overestimates the dissociation energy. B3LYP, TPSSh, and PBE0 functionals always obtain reasonable molecular geometries and dissociation energies. The overlap population density of states (OPDOS)and molecular orbital analyses indicate that the 7s, 6d, and 5f atomic orbitals of An atoms participate into the bonding of An-O bonds. QTAIM analyses have been performed to reveal the bonding nature of An-O bonds. Our results indicate that An-O bonds are essentially closed-shell characteristic. Meanwhile, we also note that An-O bonds also show partial covalent nature revealed by the high electron density and negative electron energy density.From BkO to NoO,the bond strengths are gradually decreased.

- Chinese Physics B的其它文章
- Nonlocal advantage of quantum coherence in a dephasing channel with memory?
- New DDSCR structure with high holding voltage for robust ESD applications?
- Nonlinear photoncurrent in transition metal dichalcogenide with warping term under illuminating of light?
- Modeling and analysis of car-following behavior considering backward-looking effect?
- DFT study of solvation of Li+/Na+in fluoroethylene carbonate/vinylene carbonate/ethylene sulfite solvents for lithium/sodium-based battery?
- Multi-layer structures including zigzag sculptured thin films for corrosion protection of AISI 304 stainless steel?