細菌特征分析的革蘭氏陰陽性判別算法
2021-04-12 10:13:50張勁松
小型微型計算機系統 2021年4期
關鍵詞:實驗
袁 健,趙 樺,張 明,張勁松
1(上海理工大學 光電信息與計算機工程學院,上海 200093) 2(海軍軍醫大學 教研保障中心,上海 200433) 3(中國科學院 上海生命科學研究院,上海 200031)
1 引 言
自然界存在多種多樣的病菌,如何有效地將人類新發現的細菌快速加以鑒別、分類,以便選擇有效藥物進行治療,在生物醫學領域具有重要意義.革蘭氏染色法用于鑒別細菌[1],可以把眾多的細菌分為兩大類,革蘭氏陽性菌和革蘭氏陰性菌[2].大多數化膿性球菌屬于革蘭氏陽性菌,它們能產生外毒素使人致病,而大多數腸道菌屬于革蘭氏陰性菌,它們產生內毒素,靠內毒素使人致病.在治療上,大多數革蘭氏陽性菌都對青霉素敏感,而革蘭氏陰性菌則對青霉素不敏感,卻對鏈霉素、氯霉素等敏感.所以區分出病原菌是革蘭氏陽性菌還是陰性菌,在選擇抗生素方面意義重大.
目前細菌分類方法主要是革蘭氏染色法.然而,染色時會發現某些革蘭氏陽性菌褪色,某些革蘭氏陰性菌會由于菌齡或培養基的不同而產生黑色的染色粒,同時染色程序較為復雜,由于細胞培養時間過長可能導致部分細胞發生死亡或自溶,從而導致染色結果為假陰性.革蘭氏染色法借助細菌不同的細胞壁結構引起的染色性差異來進行分類,但是涂片的厚薄和脫色時間的掌握制約著該方法的準確性,這已成為未知細菌的準確和快速分類的瓶頸.隨著第3代測序技術和質譜技術的成熟,大家已能夠很方便和快速地獲得細菌的蛋白質序列.因此,本文開創性地研究了利用計算機對細菌的蛋白質序列進行特征分析和提取來進行細菌的革蘭氏陰陽性判別的算法,經實驗證明效果良好.
本文主要完成以下3項工作:
1)提出利用細菌蛋白質序列進行細菌的革蘭氏陰陽性判別算法GCBPS算法;
2)用實驗驗證選用閉合鄰接序列模式(FCloConSP)的GCBPS算法進行細菌革蘭氏陰陽性判別的準確性以及可行性;
3)用FConSP替代GCBPS算法中的FCloConSP后生成GCBPS-X算法,比較GCBPS和GCBPS-X的準確性,以及FCloConSP相比FConSP的精簡性,驗證GCBPS算法的優化性.
2 相關工作
目前對細菌的分類方法主要有以下幾種,其中由丹麥醫生革蘭于1884年發明的革蘭氏染色法為主要的鑒別染色法[1].革蘭氏染色法根據細菌體內含有特殊的核蛋白質鎂鹽與多糖的復合物與燃料的吸附性進行分類,但是,該方法結果容易受許多因素的影響,比如菌齡和乙醇脫色時間對染色結果的影響.針對革蘭氏染色法操作復雜以及容易脫色的缺點,有一些可克服上述缺點的輔助方法,如氨肽酶法、吖啶橙染色法.此外還有利用氫氧化鉀溶液對細菌進行分類[3],此類輔助方法相比革蘭氏染色法而言操作更加簡便,時間較快.此外,基于聲光可調濾光片(AOTF)的高光譜顯微鏡成像(HMI)方法具有從細胞水平上快速鑒定微菌落中食源性致病細菌的潛力,文獻[4]利用高光譜顯微鏡成像方法對革蘭氏陽性和革蘭氏陰性食源性致病菌進行分類以及文獻[5]利用利用拉曼光譜法對革蘭氏陰陽性細胞結構所接受的拉曼散射強度不同來進行細菌對革蘭氏陰陽性判別.鑒于序列特征研究的廣泛應用[6],為了更快速、方便地實現細菌的革蘭氏陰陽性判別,本文研究對細菌的蛋白質序列進行智能分析來判別其革蘭氏陰陽性的算法.
近年來,利用序列來分類在很多領域應用頗多,尤其在基因組研究中引起了廣泛的關注[7,8],比如,利用樸素貝葉斯對rRNA序列進行分類[9]歸到Bergey的《原核生物分類大綱》.在一條生物序列中,每一項(核酸或氨基酸)都有著不同的關系,并不同以往的頻繁項集與關聯規則中的項出現的順序[10],這種序列分析工作又被稱為序列模式挖掘,主要研究如何有效地發現序列中能代表核心特征的一般序列模式(General Sequential Pattern)或精簡序列模式(Compact Sequential Pattern)[11,12].由于,精簡序列模式可以產生相對少量但分類效果、信息承載能力與一般序列模式相當的序列模式[13,14].所以本文采用精簡序列模式來分析蛋白質序列.
在精簡序列模式分析下,本文提出了基于蛋白質序列特征分析的細菌革蘭氏陰陽性判別算法(Gram Classification algorithm for Bacteria based on Protein Sequences,GCBPS),從而實現對蛋白質序列進行精簡序列模式的挖掘和特征的提取以及對革蘭氏陰陽性的判別.此算法僅需對細菌的蛋白質序列進行計算機軟件處理,無需再進行生物實驗.該方法對硬件條件要求低,判別時間短,準確性較高.
3 相關定義與問題陳述
3.1 相關定義
定義1.(鄰接子序列)
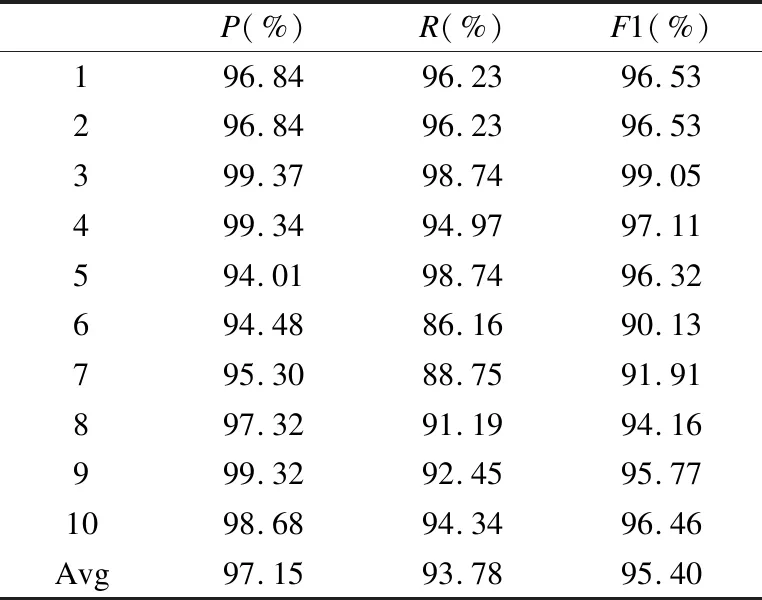
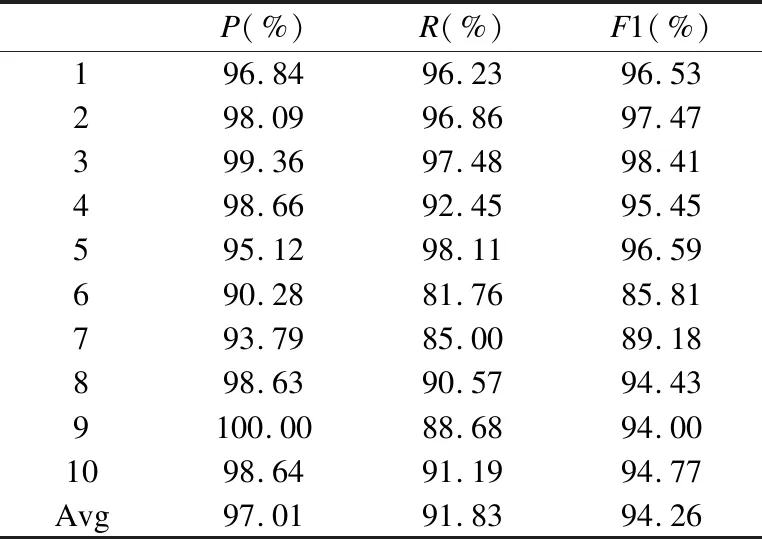
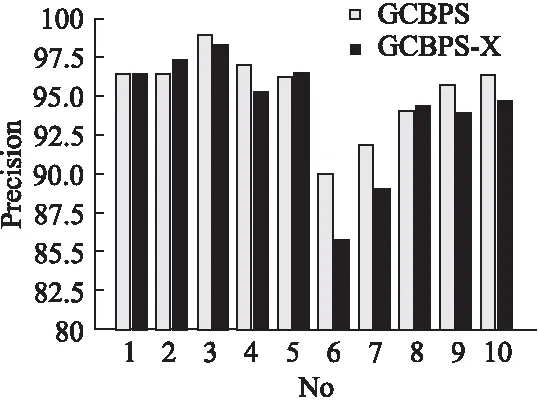
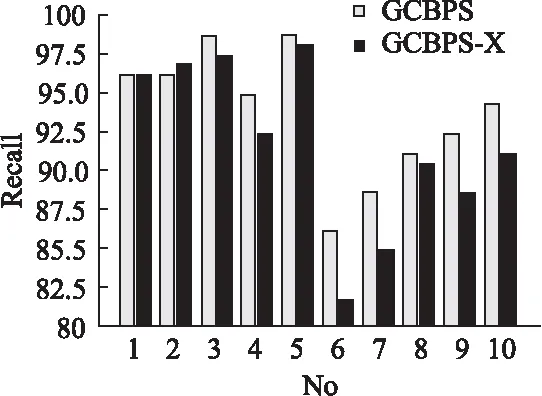
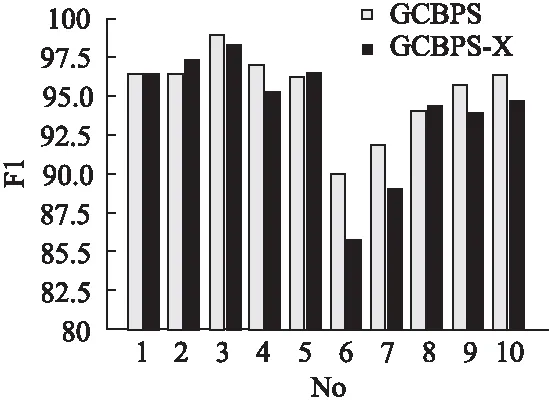
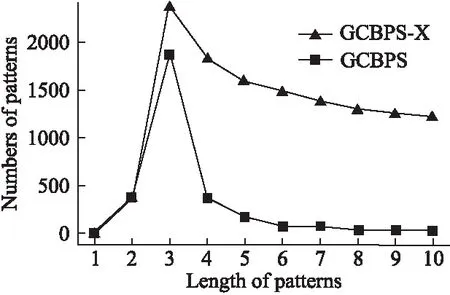
若序列S1= 定義2.(鄰接序列模式) 指定一個支持度閾值σ,若一個鄰接子序列s滿足SupD(s)≥σ,其中SupD(s)表示s支持度,則s為鄰接序列模式. 定義3.(閉合鄰接模式) 若一個鄰接序列模式s滿足不存在一個鄰接序列模式s1,同時使s?s1和SupD(s)=SupD(s1)成立,則s為閉合鄰接序列模式. 定義4.(前后子序列) 給定兩個序列s1= 需解決的問題如下: 采用第3代測序技術和質譜技術可輕松得到細菌的蛋白質序列,因此先把若干已知陰陽性的細菌蛋白質序列組成序列數據庫Seq-D,如表1中1條蛋白質短序列組成的序列數據庫Seq-D所示(第1列為序列的ID,第2列為某細菌的蛋白質序列.示例中的序列由A、B、C,3種不同項(核酸或氨基酸)組成,長度為13.)然后挖掘Seq-D中的精簡序列模式,找出細菌的革蘭氏陰陽性判別的特征,并提出細菌的革蘭氏陰陽性判別算法. 表1 含一個序列的數據庫Seq-D樣例Table 1 An example sequence database Seq-D 精簡序列模式又分為頻繁模式(FSP)、鄰接序列模式(FConSP)、閉合鄰接模式(FCloConSP)3種.若設定支持度σ為2,分別用上述3種模式對表1中的序列進行挖掘,結果如表2所示.其中第2列中,具體的模式項以及模式對應的支持度以“:”分隔,各模式項間用“,”分隔,其FSP有17個模式項,FConSP有7個模式項,而FCloConSP有5個,由以上結果可見,同一支持度下,FSP的模式項數目最多,FCloConSP的模式項數目最少. 由一般經驗可知,3種序列模式中,所包含的模式項數目越多其保留的特征也越多,故應選擇FSP來分析.但是從表2可看出FSP的模式項數目遠大于其它兩種模式,而表2所分析的序列只包含了3種氨基酸,長度只有13,實際的蛋白質序列的氨基酸可多達20種,一個序列長度可能長達上千.可想而知,選擇FSP分析序列,后續的計算量巨大,這不是一種好的選擇.因此,若能保證正確率的情況下,選擇更精簡和有效的模式,即性價比更高的序列模式,其處理時間短,更具有實用價值.故考慮從FConSP或FCloConSP產生的模式項中尋找特征完成判別.FConSP的數據量大于FCloConSP,若FCloConSP數據無法支撐準確性,則需考慮FConSP.若FConSP和FCloConSP均能保證準確性,則選擇FCloConSP更優.本文經過大量實驗,最終設計了使用FCloConSP模式的數據分析的判別算法. 表2 3種序列模式對比Table 2 Comparison of three sequential patterns GCBPS算法的流程圖如圖1所示. 該算法先對給定的已知陰陽性的序列數據庫Seq-D進行數據預處理,將Seq-D中的序列處理為特定的數據結構(S.id,S),然后針對蛋白質序列分析的特點改進了閉合鄰接模式的挖掘算法CCSpan,對訓練集數據庫中每條序列通過候選集生成、剪枝操作、閉合性篩選來挖掘FCloConSP,可以分別累計得到陰性的訓練集閉合鄰接模式特征集合和陽性的訓練集閉合鄰接模式特征集合.接著對陽性訓練集特征集合進行標準化和向量化,得到陽性特征向量. 對待測蛋白質序列進行挖掘FConSP,得到待測鄰接序列模式特征集合,再經過向量化處理得到待測序列特征向量.先計算待測向量與陽性特征向量的相似度,結果若在區間[0.8,1],則待測序列為陽性.若相似性結果不在此區間,則初步判定為陰性,其實這些序列并不一定全是陰性,還存在假陰性(陽性).因此,需進行去假陰性處理.經實驗發現直接把待測序列向量與前述方法得出的陰性特征向量比對,其正確率受限,因此經過大量試驗后修正了陰性特征庫,即把原求出的陰性特征集中長度為2的模式項去掉作為修正的陰性特征集合,再進行標準化和向量化,得到陰性特征向量.將非陽性待測向量與陰性特征向量進行相似度計算,若相似性結果在區間[0.8,1],則為陰性序列,否則為陽性序列.由此可得出最終的陰陽性判定結果. 圖1 GCBPS算法流程圖Fig.1 GCBPS algorithm flowchart CCSpan算法[15]用于挖掘一個序列數據庫的指定支持度模式集合,GCBPS算法中的序列模式挖掘部分引入了CCSpan算法的主要思想,與原CCSpan算法不同的是,GCBPS算法只挖掘單條序列的FCloConSP,更有利于保持源序列庫中每條序列的特征.在取得Seq-D中每條序列的FCloConSP后,依次輸入該序列數據庫中的下一條序列繼續挖掘,直至該序列數據庫循環結束.此算法設計了以下幾種特殊的數據結構,便于實驗計算: 1.作為輸入的序列數據庫Seq-D由一個二元結構(S.id,S)組成,其中S.id為此序列的ID編號,S則為序列本身. 2.閉合鄰接序列模式與非閉合鄰接序列模式以一個三元結構組成(s,s.count,B),其中f表示模式,s.count表示該模式在序列數據庫D上的頻數,即實際支持度,而B中有兩種值:“Y”代表模式閉合,“N”代表非閉合模式. 3.一條序列F可以分割成若干個不相交的子集合,即{{F1},{F2},…,{Fn}},其中n是最大的模式長度,F中每個子集僅僅包含單一長度(n)的模式. 本文通過以下3步來實現FCloConSP的挖掘: Step 1.取數據庫Seq-D的每一個序列S(S.id,S)按照設定的切分長度切分成一系列的片段,這些片段中所有的項均保持原有的順序和鄰接屬性.初始切分長度為2,當一輪切分片段結束后,再把上一輪切分長度+1進行下一輪切分,一直到切分長度等于原始序列長度時,切分結束.得到的片段為候選片段.此時切分序列產生的集合為{{F1},{F2},…,{Fn}},其中每個子片段為鄰接序列模式,其結構為(s,s.count,B). Step 2.采用CCSpan算法中的剪枝方法(前后子序列剪枝、支持度剪枝)對Step 1產生的候選片段進行剪枝,刪除已經出現過的片段和不滿足支持度要求(s.count<σ)的片段.經剪枝后的候選片段仍為鄰接序列模式. Step 3.對Step 2得到的鄰接序列模式進行閉合性檢查[15],則篩選出所有的非閉合鄰接序列模式并標識即(s,s.count,B)中B標識為“N”,從而以B=“Y”可篩選出該序列的閉合鄰接序列模式. Step 4.取數據庫Seq-D中的下一條序列重復Step 1-Step 3得到該條序列的FCloConSP.將所有的上述序列的FCloConSP按照已知的陰陽性放入陰性特征庫與陽性特征庫.若陽性特征庫內或陰性特征庫內中有若干相同的模式項s時,則把s.count進行累加后合并為一個模式項.合并處理后每個模式項只出現1次.最終的結果就是陰性訓練集特征庫和陽性訓練集特征庫.記為: LCloConSPs=[s1:s1.count,s2:s2.count,s3:s3.count,…,sn:sn.count]. 為實現算法的軟件編程,此部分設計了以下幾個函數: 1)函數snip():作用為獲得所有長度為1的頻繁模式,該結果用于得到長度為2的頻繁模式.在F1中每一個候選子序列實際支持度都不小于給定的閾值σ,其中每個模式都以三元組(s,s.count,Y)形式表示,標記Y為默認值. 2)函數ConSP-snip():其作用為存儲當下長度的所有模式片段的Pn作為輸入,以挖掘長度大于等于2的鄰接序列模式.經過3步剪枝操作,Pn會不斷更新已檢測片段. 3)函數CloConSP-snip():其作用為得到最后所需的閉合鄰接序列三元組.根據3.1定義中閉合鄰接序列的定義進行閉合性檢測,可得包含閉合模式與非閉合模式的集合. 4)函數Count-Patterns():最作用為統計由FCloConSP所產生的訓練集合,其中LCloConSPs存儲陰陽性訓練集集合的FCloConSP. 4.2.1 相似度計算 余弦相似度[16]用向量空間中兩個向量夾角的余弦值作為衡量兩個個體間差異的大小.余弦值越接近1,表明夾角越接近0度,則兩個個體越相似.余弦距離更多的是從方向上區分差異,而對絕對的數值不敏感.所選用的公開數據集中革蘭氏陰性菌的蛋白質序列數量遠大于陽性菌的蛋白質序列數量,在實際中亦是如此,本文在基礎的余弦相似度公式上增加了參數k,目的是在計算過程中,通過參數k對模式的頻數進行篩選,過濾不必要的模式,降低了數據的計算時間復雜度,如公式(1)所示.經過大量的實驗計算得出,當k為每組訓練集中所有模式項的頻數的中位數時,刪掉頻數小于k的模式項后的精簡訓練集集合參加判別更準確.在訓練集中,與待測序列的鄰接序列模式取交集(若訓練集中無此模式,則頻數置為0),并以模式頻數構建向量,利用公式(1)可計算待測序列向量與該組陰(陽)性訓練集的余弦相似度. (1) 其中x,y為待比較的兩個向量. 4.2.2 陰陽性判別主要步驟 Step 1.統計得到陰陽性訓練集特征庫中的模式項頻數的中位數k,并過濾陰陽性特征庫中模式項頻數小于k的模式項,即對LCloConSP中si.count≤k(1≤i≤n)的模式刪除,從而進行標準化. Step 2.將標準化后的陽性訓練集集合與待測序列取交集,若訓練集中無此模式,則頻數置為0,并以訓練集集合以及待測序列集合中模式頻數(si.count)分別向量化,即T=[s1.count,s2.count,s3.count;…,sn.count],利用公式(1)求得待測序列與陽性訓練集向量的余弦相似度,結果若在[0.8,1]則判定為陽性. Step 3.得到第1步判別結果后,對于相似性結果在[0,0.8)的序列,會出現假陰性性狀.因此先將陰性特征訓練集集合刪除模式長度為2的模式,然后進行Step 2中標準化以及向量化得到陰性訓練集向量,最后利用公式(1)求得待測序列與陰性訓練集向量的余弦相似度,結果若在[0.8,1]則判定為陰性,否則為陽性.綜合兩步判別結果得到最終待測序列的陰陽性. 此部分的主要函數為: 函數Cosin-S():用于對向量化后的訓練集以k值進行標準化,并得到測試集與陰陽性訓練集的余弦相似度Cosine_sim.其中,TN、TP為陰陽性訓練集的向量,Ttest測試集中單條序列的向量. GCBPS算法主要由兩部分組成:1)為陰陽性訓練集與測試集模式集合挖掘;2)為測試序列與陰陽性訓練集的相似性計算. 以下為GCBPS算法偽代碼,其中:原始的序列數據庫為Seq-D,最小支持度為σ.F存儲所有的鄰接序列模式,Fn存儲長度為n的序列模式.F1存儲模式長度為1的頻繁模式.模式集合F= {s,s.count,B}|f.count≥σ}為所挖掘頻繁模式的訓練集集合.集合LCloConSPs={(s,s.count,B)|f.count≥σ}存儲序列數據庫中全部序列挖掘的FCloConSP.LTest={(s,s.count,B)|f.count≥σ}存儲一條待測序列的FConSP集合.陰性訓練集向量為TN,陽性訓練集向量為TP,測試集向量為TTest. 算法GCBPS: 輸入:由待測序列組成的序列數據庫Seq-D,以及支持度 σ 輸出:Seq-D中各序列的S.id與該序列革蘭氏陰陽性判別結果 Begin: F←φ;//以F存儲CloConSPs Fn←φ;//以Fn存儲長度為n的ConSPs F1←snip//(Seq-D,σ)//獲得1-sequences 1.for(n=2;Fn-1≠φ;n++)do 2.Pn←φ//以Pn存儲當前切分長度片段 3.foreach sequence S ∈Seq-Dand l(S)≥ ndo 4.foreach con subsequence s ∈ S and l(s)= ndo 5. ConSP-snip(Seq-D,s,Fn-1,Pn,S.id,σ);//獲得ConSPs 6.endfor 7.endfor 8.Fn-1←CloConSP-snip(Fn-1,Fn);//獲得CloConSPs 9.F←∪n-1Fn-1; 10.endfor 11.LCloConSPs,LTest←Count-Patterns(F)//獲得模式集合 12.TP←LCloConSPs//訓練集集合向量化 13.TTest←LTest//測試序列模式集合向量化 14.Cosine_sim←Cosin-S(TP,Ttest)//相似度計算 15.ifCosine_sim ∈ [0.8,1]: 16. 待測序列為陽性 17.else:TN←刪除LCloConSPs中長度為2的模式 18. Cosine_sim←Cosin-S(TN,Ttest) 19.ifCosine_sim∈[0.8,1]: 20. 待測序列為陰性 21.else:待測序列為陽性 End 為了驗證GCBPS算法的準確性、可行性及優化性,設計和完成了以下兩個實驗. 論文選取蛋白質序列公開數據集PSORTb v3.0(1)https://www.psort.org/dataset/datasetv2.html.該數據集中包含1591條革蘭氏陰性菌蛋白質序列和576條革蘭氏陽性菌蛋白質序列.本文實驗選取10折交叉驗證,即1將數據集分成10組,輪流將其中9組做訓練1組做驗證,10次所得結果均值為算法精度的估計.本實驗中,采用精準率P、召回率R、值F1-score作為實驗的主要評價指標[17],計算方法如公式(2)-公式(4)所示.其中:TP:表示測試集中正確的把陰(陽)性菌預測為陰(陽)性的序列個數;FN:表示測試集中錯誤的把陰性菌預測為陽性的序列個數;FP:表示測試集中錯誤的把陽性菌預測為陰性的序列個數.F1值為綜合度量準確率和召回率的指標. (2) (3) (4) 實驗將數據集中的1591條革蘭氏陰性菌蛋白質序列和576條革蘭氏陽性菌蛋白質序列放入1個數據庫中,再將數據集均勻分為10組,每組包含革蘭氏陽性菌約57條,革蘭氏陰性菌約159條,其中1組作為測試集,余下9組作為訓練集,依次進行10組實驗. 實驗1.驗證GCBPS算法的準確性與可行性 實驗的步驟為: Step 1.取1組序列作為測試組,從中取1條未測序列作為待測序列,剩下9組數據序列,放入GCBPS的序列數據庫Seq-D; Step 2.按GCBPS的方法判別出序列的陰陽性,即把數據帶入事先編寫好的算法程序運行得出結果; Step 3.記錄算法得出的序列陰陽性結果與實際的陰陽性結果; Step 4.若測試組的序列未測試完,則重復Step 1-Step 3.若測試完,則計算該組評估指標(P、R、F1-score),并進入Step 5; Step 5.依次更換其余9組輪流作為測試組,重復Step 1-Step 4,得到10組的評估指標,并計算平均值,如表3所示. 用GCBPS算法對細菌進行革蘭氏陰陽性判別結果的實驗評價指標如表3所示.本實驗在支持度σ=2 的條件下,分別從10組實驗的精確率、召回率以及F1值來判斷該算法的準確性及可行性. 表3 GCBPS算法10組實驗評價指標Table 3 GCBPS algorithm 10 groups of experimental evaluation indicators F1是綜合度量準確率和召回率的指標,由表3可看出第3組實驗F1值最高為99.05%,10組的平均F1值為95.40%,所以GCBPS算法判別細菌的革蘭氏陰陽性的結果較準確.因此可以得出:不進行生物實驗,直接采用實現GCBPS算法的計算機軟件進行細菌的革蘭氏陰陽性判別方法是準確的和可行的. 實驗2.驗證GCBPS中選擇FCloConSP的精簡性與優化性 本組實驗選取支持度σ=2,用FConSP替代GCBPS算法中對訓練集進行特征提取的步驟,其余步驟相同,為以示區別,后稱為GCBPS-X算法. 其實驗步驟為: Step 1.按實驗1的方法步驟并跳過5.1中的Step 3后運行; Step 2.記錄GCBPS-X算法的評估結果; Step 3.統計GCBPS算法中產生的FCloConSP訓練集特征庫模式項的種類及個數. Step 4.統計GCBPS-X算法中產生的FConSP訓練集特征庫模式項的種類及個數. 由實驗可得FConSP下的10組實驗的評價指標由表4可見,圖2-圖4分別為GCBPS算法與GCBPS-X算法兩種模式準確率、召回率、F1值對比. 由表3可知,GCBPS判別實驗F1值均值為95.40%;由表4可知,GCBPS-X判別實驗F1值均值為94.26%.因此,GCBPS算法比GCBPS-X算法綜合準確率高.由圖2可知有6組實驗的準確率是GCBPS高于GCBPS-X;由圖3可知有8組實驗的召回率是GCBPS高于GCBPS-X;由圖4可知有6組實驗的F1值是GCBPS高于GCBPS-X,由此可見GCBPS比GCBPS-X的判別準確率更高. 表4 GCBPS-X算法10組實驗評價指標Table 4 GCBPS-X algorithm 10 groups ofexperimental evaluation indicators 接下來進行兩種算法中模式項的精簡性對比.圖5為革蘭氏陰性菌GCBPS與GCBPS-X兩種算法中模式項數目的對比圖,其中橫坐標為模式項的長度,縱坐標為模式項的數目.在陰性菌序列數據庫中,FConSP共有130978個模式項,FCloConSP有11064個模式項,由此可見,FCloConSP的數目遠遠小于FConSP.由圖5可以看出兩條曲線在同一支持度下(σ = 2)呈下降趨勢,FCloConSP的模式項主要集中在長度為3和4之間,分別占比46.74%與39.34%,而在FConSP中,長度為3與長度為4的模式僅占4.09%與4.88%.隨著模式長度增長的同時,FCloConSP中模式長度較長的模式為0.相比而言,GCBPS中的FCloConSP更為精簡. 圖6為革蘭氏陽性菌GCBPS與GCBPS-X兩種算法中模式項數目的對比圖,其中橫坐標為模式項的長度,縱坐標為模式項的數目.在陽性菌576條序列數據庫中,FConSP數目為203494條,而FCloConSP僅為3323條,由此可見,FCloConSP的數目遠遠小于FConSP.在FCloConSP中,長度為3和4的模式數目分別為1869與360,分別占比為56.24%與10.83%,而在FConSP中,模式長度為3和4的數目為2376與1841,分別占比為1.17%和0.90%.由此可見,在同一支持度下,FCloConSP的模式長度小于普通FConSP;FCloConSP集合大小遠遠小于FConSP集合. 實驗結果表明,GCBPS算法選擇FCloConSP進行序列特征分析,所處理的模式項數目更少,軟件運行時間更短,準確性更高,其具有精簡性和優化性的特點. 圖2 算法準確率比較Fig.2 Comparison of algorithm precision 圖3 算法召回率比較Fig.3 Comparison of algorithm recall 圖4 算法F1值比較Fig.4 Comparison of algorithm F1 value 圖5 革蘭氏陰性菌特征集兩種模式項數目對比Fig.5 Comparison of the number of two model items in the Gram-negative bacterial feature sct 圖6 革蘭氏陽性菌特征集兩種模式項數目對比Fig.6 Comparison of the number of two model items in the Gram-positive bacterial feature set 本文首次提出的用計算機軟件實現的GCBPS算法是細菌革蘭氏陰陽性判別領域的創新方法.算法通過對已知陰陽性序列的挖掘和分析,提取出陰性和陽性序列的特征向量,再將待測未知陰陽性序列進行相似性判別,可得出陰陽性結果.該方法中的陰陽性特征向量可以通過不斷增加數據庫中已知陰陽性序列的數量來進行動態更新,從而可持續提高判別準確性.為了縮短判別時間,也可事先運行軟件的訓練集訓練部分,判別時直接運行待測序列與前述的訓練結果比對判別的部分即可. 在未來的工作中,將進一步優化序列特征提取時所選用的模式,盡可能減少丟失的特征,提高判別準確性和縮短計算時間.以后還將嘗試把GCBPS算法用于亞細胞的定位.3.2 問題陳述


4 GCBPS算法
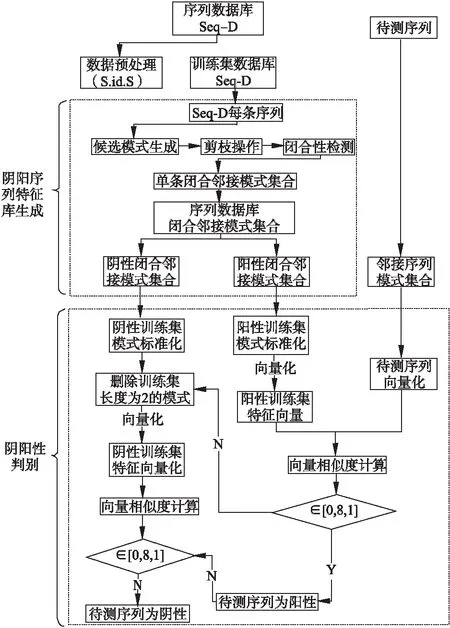
4.1 陰陽序列特征庫生成
4.2 陰陽相似性判別
4.3 算法過程
5 實 驗
5.1 實驗設置
5.2 實驗及結果分析

6 總 結
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
