中醫藥治療肌萎縮側索硬化療效機制的研究策略
2021-04-14 00:34:14吳曉俊
神經病學與神經康復學雜志 2021年2期
潘 昊,劉 特,吳曉俊
1.重慶醫科大學藥學院,重慶 400016
2.上海市中醫老年醫學研究所,上海 200031
3.上海中醫藥大學中藥研究所,上海 201203
肌萎縮側索硬化(amyotrophic lateral sclerosis,ALS)是以脊髓前角細胞、腦干運動神經核以及錐體束受累為主要表現的運動神經元病,其病情進展迅速、預后極差,通常在發病后2~5 年因多器官衰竭而死亡[1],因此也是一種具有致命性的神經退行性疾病。絕大多數的ALS為散發性,家族性ALS 約占5%。目前尚無確切有效的針對ALS 的治療藥物。
無論是散發性還是家族性ALS,因其表現為漸進性的肌肉痿軟無力甚至萎縮,因此中醫認為其屬于“痿癥”。大多數的中醫醫家認為本病以本虛為主或虛實夾雜,其中本虛以脾腎陽虛為主,夾雜胃、肝和肺虧虛。因此,目前中醫大多使用健脾益氣補腎法治療ALS,取得了一定的療效[2]。然而,已發表的ALS 中醫臨床研究大多為個案報道或病例數較少,并且研究設計欠規范,也缺少機制研究,因此較難獲得廣泛的認可,對ALS治療的指導意義有限,極大地阻礙了中醫藥診治ALS 研究的發展。此外,既往研究建立的ALS 模型系統及治療策略既有優勢,也存在不足[3]。
潘衛東團隊與朱旭瑩團隊基于中醫藥治療ALS的確切療效,使用模擬散發性ALS 進程的條件性敲除ADAR2 基因的小鼠(AR2 小鼠)以及能夠反映家族性ALS 發病機制的SOD1G93A動物模型,對中醫藥治療ALS 的機制進行了有益的探索[4-5]。本文對ALS 發病機制的最新進展進行綜述,并且結合中醫藥治療ALS 基礎研究的新進展,提出探索中醫藥治療ALS 療效機制的研究策略,以期為中醫藥治療神經退行性疾病的研究提供更多的理論依據。
1 ALS 的發病機制
既往研究認為ALS 的主要發病機制是運動神經元損傷。隨著基因、分子細胞學和轉基因動物相關研究的進展,目前認為神經膠質細胞炎癥在ALS 發病機制中發揮重要作用。運動神經元與神經膠質細胞相互作用,共同參與ALS 的發病。
1.1 神經元損傷相關發病機制
1.1.1 興奮性神經元毒性機制
ALS 的發病機制至今未明,特別是對于散發性ALS 的發病機制仍未達成共識。目前,興奮性神經元毒性和鈣離子超載被認為是ALS 神經元損傷的最主要機制之一,其中以興奮性氨基酸的細胞毒性誘發鈣離子過度內流的細胞毒性假說為代表(圖1)。
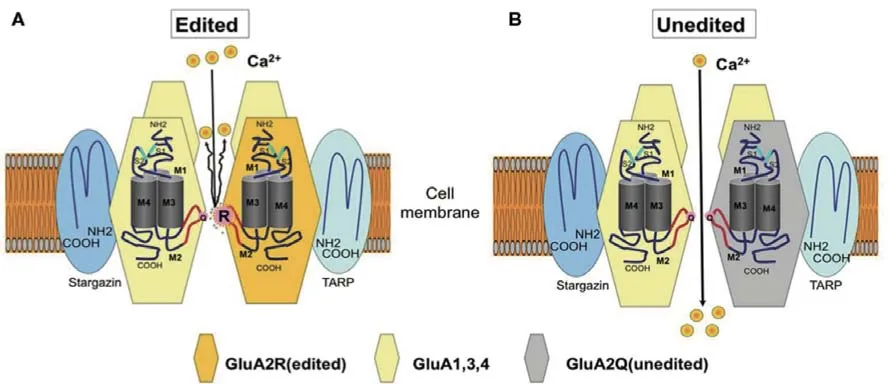
圖1 興奮性氨基酸的細胞毒性誘發鈣離子過度內流。A:正常編輯的離子型谷氨酸受體GluA2;B:條件性敲除ADAR2基因的小鼠(AR2小鼠)的運動神經元僅表達Q/R 部位未編輯的GluA2,出現異常Ca2 +滲透,從而導致神經元進行性死亡。
通過條件性敲除ADAR2 基因構建模擬ALS發病及疾病進展的AR2 小鼠模型,ADAR2 缺乏導致小鼠脊髓前角細胞發生神經變性,缺乏ADAR2 的運動神經元表達未經Q/R 位點編輯的GluA2,其亞基裝配中未經編輯的離子型谷氨酸受體GluA2 出現異常Ca2+滲透,從而導致神經元進行性死亡。此外,在AR2 小鼠中,異常的AMPA 受體導致Ca2+內流而過度激活Ca2+依賴的鈣蛋白酶(calpain);鈣蛋白酶將1 號常染色體TARDBP基因編碼的TDP-43 蛋白降解成具有聚集傾向的片段,這些片段是TDP-43 致病的基礎;激活的鈣蛋白酶還可以通過切割參與核質轉運的分子(包括核孔蛋白)破壞細胞核-細胞質轉運以及基因表達。上述證據啟發研究者針對ADAR2 基因表達下調,探索ALS 的分子靶向治療策略[6-7],并且通過使用ADAR2 基因敲除動物模型開展了一系列干預策略研究。例如,給予AR2小鼠口服吡侖帕奈或者進行以腺病毒相關病毒為載體的ADAR2 基因治療,均取得了一定的療效,但均為臨床前研究[8-9]。在開展中醫藥治療ALS療效機制的研究時,可參考上述研究方法。
1.1.2 線粒體功能障礙
線粒體對于細胞的存活和代謝具有關鍵作用,對神經元而言尤為重要。在神經元中,除了通過氧化磷酸化產生ATP 以外,線粒體在磷脂合成、鈣穩態和細胞凋亡中也具有重要作用。盡管大腦的質量只占人體質量的2%,但消耗了人體所產生的20%的ATP(靜止休息狀態),因此神經元有著很高的代謝需求。鑒于神經元的高代謝活性和能量需求,線粒體維持能量的產生以及鈣穩態對于神經元功能的維持而言尤為重要。
許多與家族性或散發性ALS 發病機制相關的蛋白如超氧化物歧化酶(superoxide dismutase,SOD)1、TDP-43、肉瘤融合蛋白(fused in sarcoma,FUS)和C9orf72 二肽重復蛋白,已被證實與線粒體之間存在相互作用[10-11]。有研究發現,ALS 患者神經元的細胞呼吸速率下降以及ATP 生成減少,此外在散發性ALS 患者的脊髓中發現電子傳遞鏈復合物Ⅰ、Ⅱ、Ⅲ和Ⅳ的活性降低[12-13]。SOD1G93A轉基因小鼠在出現ALS 運動癥狀之前,腦和脊髓中ATP 的合成已受損,線粒體呼吸速率亦下降,并且貫穿于整個病程中,同時伴有電子傳遞復合物鏈活性的下降[14]。
WIEDEMANN 等[15]發現,腦源性神經營養因子(brain-derived neurotrophic factor,BDNF)通過調控線粒體運動促進BDNF 介導的神經遞質釋放;BDNF 通過酪氨酸激酶受體B(tyrosine kinase receptor B,TrkB)激活下游PI3K、PLC-γ 和瞬時受體電位陽離子通道(transient receptor potential cation channel,TRPC)信號通路,促進細胞內鈣的釋放以及細胞外鈣離子內流,該作用在突觸部位尤為明顯;線粒體外膜上的鈣離子感受器Miro1 與鈣離子結合后,促使線粒體從微管上脫落下來,并在突觸前部位聚集,為BDNF 介導的神經遞質的釋放提供能量。中醫藥治療ALS 的療效機制中是否也涉及調控運動神經元的線粒體功能及其動態變化,有待進一步探索。
1.1.3 軸漿轉運障礙
軸漿轉運障礙可能是導致ALS 患者神經元損傷的另一個重要途徑。BDNF 選擇性地結合并激活TrkB,二者結合是逆軸漿轉運的關鍵一環。研究發現,JNK 相互作用蛋白3(JNK interacting protein 3,JIP3)可以通過與TrkB 的近膜區直接結合,將TrkB 連接至馬達蛋白kinesin-1 的輕鏈上,介導TrkB 的順軸突轉運,并且調節BDNF 誘導的細胞外調節蛋白激酶(extracellular regulated protein kinase,Erk)的激活以及軸突中絲狀偽足的形成[16]。
磷酸化的神經絲蛋白(neurofilament protein,NFP)在病理狀態下進入軸突后,NFP輕鏈(neurofilament-light,NF-L)和NFP 中鏈(neurofilament-middle,NF-M)最先開始磷酸化;隨后,NF-M 和NFP 重鏈(neurofilament-heavy,NF-H)的磷酸化作用逐漸增強,而NF-L 的磷酸化程度逐漸減弱。在NFP 的磷酸化過程中,蛋白激酶A 和蛋白激酶C 等分別作用于其亞基的不同特異性序列上,促進多種底物磷酸化。NFP 過度表達(異常磷酸化)可導致其降解障礙或發生異常聚集,嚴重影響NFP 向軸漿運輸,引發神經元變性,從而導致ALS。
上述研究闡述了SOD1G93A轉基因小鼠的脊髓前角運動神經元核周出現NFP 的異常磷酸化以及發生聚集的原因。通過基因調控抑制SOD1G93A轉基因小鼠運動神經元核周NFP 的異常磷酸化,減少其在核周的異常聚集,維持細胞骨架的完整性,以及改善軸漿運輸,從而延緩神經元變性,這可能也是中醫藥治療ALS 療效機制的研究方向之一。
1.1.4 神經營養因子缺陷
神經營養因子是一類能夠促進神經元存活和分化以及維持功能完整的蛋白質,包括BDNF、睫狀神經營養因子(ciliary neurotrophic factor,CNTF)、膠質細胞源性神經營養因子(glial cell line-derived neurotrophic factor,GDNF)、胰島素樣生長因子(insulin like-growth factor,IGF)和成纖維生長因子(fibroblast growth factor,FGF)等[17]。在這些神經營養因子中,BDNF 最受關注。BDNF 是腦內含量最豐富的神經營養因子,增強BDNF 信號已成為延緩ALS 病情進展的干預策略之一。
神經營養因子可通過旁分泌、內分泌或自分泌方式釋放。旁分泌的神經營養因子來自于鄰近細胞如膠質細胞、施萬細胞或毛細血管內皮細胞和肌纖維等[18]。內分泌的神經營養因子則來源于遠端細胞如室管膜細胞,通過血液或腦脊液進行傳遞[19]。自分泌則是指運動神經元自我提供神經營養因子的能力。
通過小分子藥物調節BDNF 受體TrkB 以增強BDNF 信號的傳遞,可以延長體外退化神經元的存活能力[20],并使ALS 模型小鼠的運動神經元丟失減少、運動能力提高[21]。BDNF 也被證實能夠增強體外培養的運動神經元的存活能力[22]。本課題組的前期研究發現,在經β-淀粉樣蛋白(amyloid β-protein,Aβ)處理的神經細胞和老年性癡呆小鼠模型中,活性異常升高的糖原合成酶激 酶3β(glycogen synthase kinase 3β,GSK3β)磷酸化發動蛋白1(dynamin-1,Dyn1)的第774 位絲氨酸,導致TrkB 內吞以及下游Akt 信號通路激活受損;使用多肽TAT-Dyn1-SpS 可以特異性阻止GSK3β 對Dyn1 磷酸化,挽救受損的TrkB 內吞以及下游Akt 信號激活,從而改善小鼠的學習和記憶能力[23]。
對于ALS 而言,神經肌肉接頭(neuromuscular junction,NMJ)間的信號傳遞可以提供新的干預靶點,這是因為ALS 中存在神經突觸末梢與肌肉之間的連接障礙。事實上,適當的合理運動在延緩ALS 病情進展、保護NMJ 以及維持運動功能以提高生活質量等方面顯示出一定的療效。BDNF基因的轉錄、翻譯和分泌受神經活動調節,而運動訓練能夠增加嚙齒動物脊髓和骨骼肌中BDNF mRNA 和蛋白的表達。在ALS大鼠模型中,大腦和跖肌中BDNF/TrkB 信號通路嚴重受損,而恢復BDNF 信號是一個很好的ALS 治療選擇,能夠通過結合TrkB 促進損傷的運動神經元存活[24]。對于恢復BDNF 信號通路的探索,可能也是中醫藥治療ALS 療效機制的研究方向之一。
1.2 神經炎癥相關發病機制
神經炎癥是ALS 的病理學表現之一。神經炎癥是由活化的小膠質細胞、星形膠質細胞以及浸潤的CD4+和CD8+T 淋巴細胞引起,與ALS 病程有關[25-26]。研究發現,神經炎癥可能由退化的運動神經元與周圍小膠質細胞之間的相互作用所引起,而小膠質細胞介導的突觸修剪可能導致ALS神經突觸的進行性丟失[27]。
與廣泛表達SOD 突變體的轉基因小鼠相比,僅在神經元中表達SOD 突變體可能無法誘發運動神經元疾病,或者運動神經元疾病的發病較晚、進展緩慢并且小鼠的生存時間較長[28-29]。另一項研究發現,在運動神經元以外的其他細胞中表達SOD 突變體的動物模型仍出現ALS 病理學改變,表明運動神經元之外的其他細胞對ALS 的致病至關重要[30-31]。并且,從ALS 動物模型的小膠質細胞中特異性去除SOD 突變體后,其病情進展明顯延緩,存活率增加[32-33]。給予尚未出現癥狀的SOD1G37R轉基因小鼠腹腔注射脂多糖(lipopolysaccharide,LPS)后可導致ALS 癥狀提前出現[34]。由此可見,小膠質細胞參與了ALS的病理生理過程。
激活的小膠質細胞至少有2 種不同的表型,即M1 型(毒性)和M2 型(保護性),以響應不同的微環境信號,進而分泌多種效應分子[35]。M1 型小膠質細胞釋放促炎癥介質,包括白細胞介素(interleukin,IL)-1、IL-6、腫瘤壞死因子、趨化因子、前列腺素E2和誘導型一氧化氮合酶等[30-31]。由抗炎細胞因子IL-4、IL-10 或IL-13誘導的M2 型小膠質細胞能夠抑制炎癥,通過吞噬作用清除細胞碎片,促進細胞外基質重建,并且通過釋放營養因子支持神經元的存活[35]。對SOD1G93A轉基因小鼠的小膠質細胞進行轉錄組分析,發現參與抗炎途徑的基因(包括IGF1、顆粒蛋白前體和髓系細胞觸發受體2 基因)與潛在的神經毒性因子(包括基質金屬蛋白酶12 和經典促炎細胞因子)相關基因的表達均上調[36],表明ALS 小鼠同時表達M1 型和M2 型小膠質細胞。
將野生型星形膠質前體細胞局部移植至SOD1 基因突變大鼠的頸脊髓,可以延緩大鼠的疾病進展,并且延長生存時間[33]。反之,將表達SOD1G93A突變體的星形膠質前體細胞移植至野生型小鼠的脊髓,可導致局部運動神經元變性,引發中度的運動功能障礙[34]。由此提示,星形膠質細胞在ALS 的病理進程中亦發揮一定的作用。
星形膠質細胞經誘導可表現為神經毒性A1型反應性星形膠質細胞或神經保護性A2 型反應性星形膠質細胞[35]。神經炎癥刺激物(如LPS)可誘導生成神經毒性A1 型反應性星形膠質細胞,引發神經毒性以及促進神經退行性變;缺血可誘導生成神經保護性A2 型反應性星形膠質細胞,后者通過分泌神經營養因子以促進神經保護和神經修復。正常的星形膠質細胞可以通過分泌某些因子(研究最多的是神經營養因子),遠距離保護ALS 患者的運動神經元。
2 基于臨床前研究制定中醫藥治療ALS 療效機制的研究策略
基于中醫理論“脾主四肢肌肉”“腎者作強之官”,ALS 的基本病機為脾腎陽虛。使用AR2小鼠模型開展中醫藥治療ALS 的療效研究,發現健脾補腎方能夠明顯改善小鼠的四肢握力,以及延緩疾病進展,推測可能是通過調控ADAR2 基因的表達而發揮作用。在SOD1G93A轉基因小鼠模型中,健脾補腎方的療效也得到了進一步的驗證,顯示健脾補腎方能夠明顯延遲ALS 的發病時間、改善運動功能以及延長生存時間;推測其作用機制可能是通過抑制NFP 異常磷酸化以及減少其在核周的聚集,從而維持細胞骨架的完整、減輕軸索的萎縮、改善軸漿運輸以及延緩神經變性,繼而達到保護神經細胞的目的[36]。
目前有不少基于中醫理論“健脾補腎”辨證論治ALS,并且取得一定療效的中藥方劑。這些方劑是否通過保護神經元細胞和(或)調節膠質細胞活性等機制而發揮治療作用,有待進一步探討。
本課題組認為,中醫藥治療ALS 療效機制的研究策略是開展中藥方劑干預不同的ALS 轉基因動物模型的療效驗證研究,旨在探索中藥方劑對于神經元和膠質細胞的作用以及其保護神經元和調節神經膠質細胞的分子機制,并在此基礎上構建轉基因小鼠以進一步驗證中藥方劑治療ALS 的機制(圖2)。通過開展上述研究,提出中藥方劑及其活性成分對于神經細胞毒性可能的作用機制,例如通過調節谷氨酸受體亞基的活性來調控病理性TDP-43 片段的聚集,改善神經元的線粒體功能及其軸漿轉運,促進神經營養因子的合成和分泌,或者在神經膠質炎癥中抑制有害炎癥以及調節抗炎活性因子等。
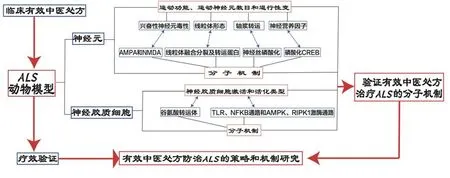
圖2 中醫藥治療肌萎縮側索硬化療效機制的研究策略
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
現代臨床醫學(2021年3期)2021-07-16 07:36:44
中國民間療法(2021年5期)2021-06-09 09:21:42
學苑創造·A版(2020年9期)2020-10-13 09:41:02
文苑(2018年21期)2018-11-09 01:23:06
知識經濟·中國直銷(2017年7期)2017-07-24 14:12:41
中國衛生(2016年11期)2016-11-12 13:29:24
中國衛生(2015年9期)2015-11-10 03:11:12
中國衛生(2014年3期)2014-11-12 13:18:12
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
