中性粒細胞胞外誘捕網在痛風性關節炎中的作用:一枚硬幣的兩面*
2021-04-29 01:05:12王雪霖曹秀梅閆建設
自然雜志 2021年2期
王雪霖,曹秀梅,閆建設?
①上海大學 生命科學學院,上海 200444;②上海交通大學 醫學院,上海市免疫學研究所,上海 200025
痛風性關節炎(gouty arthritis, GA)是由單鈉尿酸鹽(monosodium urate, MSU)晶體沉積引發的炎癥性疾病[1]。當痛風性關節炎患者的尿酸濃度超過尿酸的溶解度達到過飽和狀態時,MSU晶體會在軟骨、滑膜及周圍組織沉積并刺激關節滑膜,發生一系列病理反應,從而引發關節處出現急性炎癥[2]。此時,血管內皮表面的黏附分子會捕獲循環血中的中性粒細胞,滲出血管,定向遷移至關節腔的炎癥部位,識別并對抗入侵宿主體內的病原體,發揮保護作用[3]。近年研究發現,除了吞噬及脫顆粒作用外,中性粒細胞也可以通過形成中性粒細胞胞外誘捕網(neutrophil extracellular traps,NETs)的方式消滅入侵的病原微生物[4]。NETs由于吞噬刺激從中性粒細胞釋放的胞外DNA纖維,幫助嗜中性粒細胞固定并誘捕細菌、真菌或病毒,從而更有效地消除這些病原體[5]。Brinkmann等人[6]率先發現NETs,他們通過免疫熒光及DNA染料染色的方法對NETs的組成進行分析后得出,該結構是由染色質和蛋白顆粒組成的、能捕獲并殺死微生物的胞外網狀結構。MSU晶體誘發中性粒細胞NETs產生,后者反過來吞噬捕獲MSU晶體,并通過鑲嵌在NETs上的彈性蛋白酶、髓過氧化物酶、組織蛋白酶G等酶類的殺菌作用,發揮其抗菌作用,防止炎癥擴散[7]。
鑒于中性粒細胞NETs在GA中發揮的重要作用,本文將對GA的發生、發展及炎癥消散過程進行概述,重點討論中性粒細胞NETs在GA中所起到的雙重作用,并對GA中參與中性粒細胞NETs形成的相關信號通路進行簡述。
1 痛風性關節炎
1.1 痛風性關節炎發生及自我緩解
痛風性關節炎疾病發作過程主要包括急性炎癥反應和炎癥自行消散兩個階段。痛風是由關節內和關節周圍沉積的MSU晶體引發的、具有典型急性炎癥特征的疾病,主要表現為嚴重的關節疼痛、腫脹、紅斑和功能喪失。當血尿酸濃度超過正常閾值(成年男性237.9~356.9 μmol/L,女性178.4~297.4 μmol/L)時,機體會形成高尿酸血癥。部分患者體內過量的尿酸以晶體形式析出并沉積于關節滑膜、軟骨以及周圍其他組織,引發一系列炎癥反應,進而誘發GA。GA的一個顯著特征是其具有“自限性”,患者即使未經治療,且在其炎性部位仍然可以檢測到MSU的情況下,其癥狀亦可在7至10天內得到緩解。在關節處沉積的MSU晶體破壞了機體正常穩態平衡,引發炎癥反應。機體為維持原有穩態會調動自身免疫應答機制,包括中性粒細胞產生NETs對晶體進行包埋,暫時性抑制炎癥擴散[8]。
1.2 痛風性關節炎相關病理機制
痛風性關節炎患者滑液和關節周圍的MSU晶體被先天免疫系統識別為異物,引發機體免疫反應。MSU晶體激活單核巨噬細胞中Toll樣受體(TLR)和NALP3炎性小體的功能[9],隨后活化半胱氨酸蛋白酶-1(caspase-1)。后者將已經存在于細胞中的白介素-1β(IL-1β)的無活性前體和白介素-8(IL-8)轉化為有活性的成熟細胞因子,并迅速釋放至胞外[2]。研究表明,IL-1β和IL-8激活一些促炎相關信號轉導通路,包括NF-κB和MAPK通路。其中,NF-κB不僅能夠影響促炎細胞因子(比如IL-1、IL-18)、腫瘤壞死因子(TNF-α)和其他炎性介質的轉錄,還會影響粒細胞趨化因子(IL-8)的轉錄[10]。促炎因子和細胞趨化因子的大量產生,導致中性粒細胞浸潤加劇[1],產生痛風發病中的炎癥放大效應(圖1)。
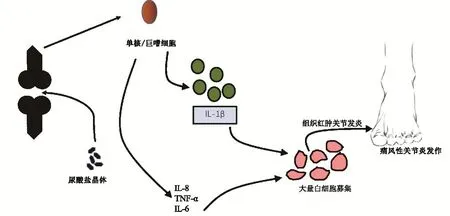
圖1 痛風性關節炎發病過程
痛風性關節炎自限性的形成機制存在多種假說。有研究指出,痛風性關節炎發作早期,肥大細胞(mast cell, MC)與巨噬細胞早于中性粒細胞活化,并釋放前列腺素、組胺等多種細胞因子和趨化因子刺激腺體分泌、增加毛細血管通透性,利于中性粒細胞通過毛細血管壁遷移至炎癥部位。當大量中性粒細胞在關節處聚集時,可通過釋放胞外誘捕網對晶體進行吞噬包埋,減緩炎癥進程,使得炎癥暫時消散[11]。還有研究指出,在痛風性炎癥反應中發揮關鍵作用的NLRP3炎癥小體活化需要上游胞質蛋白、下游caspase-1前體以及銜接蛋白ASC激活,一些胞內炎癥小體調節劑(如COPs、POPs等)可通過調節NLRP3炎癥小體活性,競爭性結合銜接蛋白ASC,阻斷成熟IL-1β釋放,抑制炎癥反應發生[12]。此外,也有研究指出,內源性IL-1受體拮抗劑(interleukin-1 receptor antagonist, IL-1Ra)通過競爭IL-1受體,阻斷由MSU晶體刺激免疫細胞釋放的IL-1促炎活性,抑制炎癥反應[8]。痛風性關節炎中炎癥消散能夠抑制輻射性炎癥發生,是機體為維持內環境穩態所進行的自我調節過程。
2 中性粒細胞NETs在痛風性關節炎中的雙重作用
2.1 中性粒細胞NETs促進痛風性關節炎的炎癥反應
在痛風性關節炎中,MSU晶體刺激產生的大量IL-1β在與中性粒細胞及巨噬細胞受體結合之后,對接頭蛋白MyD88進行激活。Liu-Bryan等人[13]的研究表明,單核細胞中表達的TLR- 2、TLR-4和它們共享的接頭蛋白MyD88是促進中性粒細胞活化、啟動機體免疫反應主要的決定因素之一。MyD88蛋白的激活引發核轉錄因子NF-κB活化,NF-κB又能引發與炎癥免疫反應中密切相關的炎癥因子(如IL-8、TNF-α)以及一些刺激炎癥反應產生的酶(如環氧合酶-2(COX-2)、磷脂酶等)表達,誘使眾多中性粒細胞浸潤至關節處使炎癥反應加劇[14]。中性粒細胞被大量募集至關節處時,通過產生NETs這一胞外網狀結構發揮其功能。痛風性關節炎中MSU晶體在組織和關節處沉積是引發炎癥部位NETs形成的主要原因。中性粒細胞在對MSU晶體攝取的同時,粒細胞和單核細胞會釋放更多促炎細胞因子和趨化因子,從而促進炎癥反應[15]。
2.2 中性粒細胞NETs抑制痛風性關節炎的炎癥反應
Mitroulis等人[16]的研究首次發現中性粒細胞NETs在痛風性關節炎的炎癥消散中具有重要作用。隨后他們發現在使用IL-1抑制劑處理后,中性粒細胞形成NETs的能力會明顯下降,表明NETs形成過程與IL-1的激活有密切關系[17]。此外,中性粒細胞對MSU晶體的攝取導致自身釋放大量細胞因子,包括炎癥介質(如TNF-α和IL-6)、中性粒細胞引誘劑(如IL-8)和激活劑(如CCL3和CXCL10),引發NETs大量形成聚集型NET(aggregated NET, aggNET)。aggNET通過降解細胞因子和趨化因子并破壞嗜中性粒細胞的募集和活化,促進痛風性關節炎的炎癥消退[18]。由此可見,NETs形成在痛風性關節炎的炎癥消散中起著至關重要的作用。
痛風性關節炎發展至晚期,會在關節等處出現痛風石。痛風石會逐漸浸入到骨質中,這是痛風性關節炎患者關節處出現骨破壞的主要原因[19]。研究表明,痛風石的組成成分和NETs十分相似。在顯微鏡下觀察,痛風石表現為慢性肉芽腫性損害,主要包括由致密結締組織圍繞尿酸單鈉化合物晶體為核心所形成的單核/多核巨噬細胞集合物[20]。當發炎部位出現中性粒細胞密度很高的情況時,NETs形成的聚集體aggNET會密集地包裹MSU晶體,通過自身所含蛋白酶捕獲并降解一些促炎介質。Chatfield等人[21]的研究表明NETs在痛風的急性炎癥以及未發炎的痛風石周圍都存在,且其對DNase具有一定抗性,因而在組織中它可通過由DNA包覆的聚集體將晶體隔離,并對發生的炎癥反應進行抑制。研究發現,痛風石中同時存在抗炎因子如轉化生長因子-β(transforming growth factor-β, TGF-β)和促炎因子例如IL-1的表達,導致痛風性關節炎中存在慢性炎癥周期,癥狀為不斷反復發作和自我緩解[22]。
由此可見,在痛風性關節炎中中性粒細胞NETs的形成具有雙重作用:一方面,NETs形成過多造成聚集后,就易形成痛風石造成骨質侵蝕并誘發機體的慢性炎癥反應;另一方面,NETs能夠對MSU晶體進行包埋和清理,保護機體。
3 調控痛風性關節炎中NETs形成的相關信號通路
在痛風性關節炎中,MSU 晶體在關節組織處沉積會引起關節處的免疫細胞釋放一些促炎性的細胞因子和趨化因子(如IL-1β、TNF-α、IL-8等),進而誘導大量的中性粒細胞浸潤至關節腔,刺激中性粒細胞激活,引發嚴重的炎癥反應。近年的研究表明,痛風性關節炎發病機制受許多因素,例如一些信號通路(ERK1/2、p38 MAPK)、信號傳導(炎性細胞因子及Src家族PTK)等影響。MSU晶體通過Src家族的PTK信號傳導,誘導單核巨嗜細胞中IL-8啟動子的活化和IL-8的表達[23]。在IL-8、IL-1β等炎性細胞因子的作用下,中性粒細胞大量募集,促進炎癥反應的進程。此外,MSU晶體也誘使補體激活,通過產生具有趨化作用的C5a促進中性粒細胞浸潤,從而影響痛風性炎癥反應[24]。大量研究顯示,中性粒細胞浸潤到關節腔所產生的炎癥反應是痛風性關節炎病理機制的核心,下文就IL-8和IL-1β對中性粒細胞趨化性及NETs形成的影響進行討論。
3.1 IL-1β信號通路
痛風性關節炎發病機制較為復雜,有許多與中性粒細胞NETs形成相關的炎性細胞因子及信號通路參與調控。研究表明,IL-1β信號通路對痛風性關節炎中NETs的形成具有重要作用。IL-1β具有多種功能,它是炎性反應的重要介質,主要由單核巨噬細胞產生,參與多種細胞活動,包括細胞增殖、分化和凋亡等生理活動[17]。IL-1β產生至少受到兩個不同步驟的嚴格控制:首先,IL-1β蛋白前體的產生,當MSU晶體作為一種異物被細胞表面和胞質模式識別受體(TLR2、TLR4、CD14等分子)識別后,引發巨噬細胞內IL-1β轉錄啟動子的激活,促進IL-1β前體生成[25];隨后,對IL-1β前體進行切割以產生具有活性的IL-1β蛋白,對IL-1β前體的加工涉及到上游信號caspase-1的激活,有活性的caspase-1能夠對其加工剪切[9]。caspase-1的激活需要上游NALP3炎性體激活,在痛風性關節炎中通過沉積的MSU晶體對NALP3炎性體激活,從而誘導IL-1β釋放(圖2)[25]。NLRP3廣泛表達于樹突細胞、單核細胞和巨噬細胞,具有識別病原體的功能。ASC是NLRP3炎性體中的銜接蛋白,將NLRP3炎性小體和caspase-1前體聯系起來,形成具有酶活性的異二聚體caspase-1。活化的炎性體效應蛋白caspase-1,將無活性的IL-18和IL-1β前體剪切為成熟的IL-18和IL-1β并釋放到細胞外。IL-1β與滑膜細胞、成纖維細胞、內皮細胞等細胞表面的IL-1β受體相互識別,激活接頭蛋白MyD88,使NF-κB功能激活,引發炎性細胞因子基因表達,產生包括IL-1、IL-6、TNF-α和中性粒細胞的趨化因子等,促使中性粒細胞進入晶體沉積部位發揮免疫功能,并形成胞外誘捕網吞噬MSU晶體[26]。
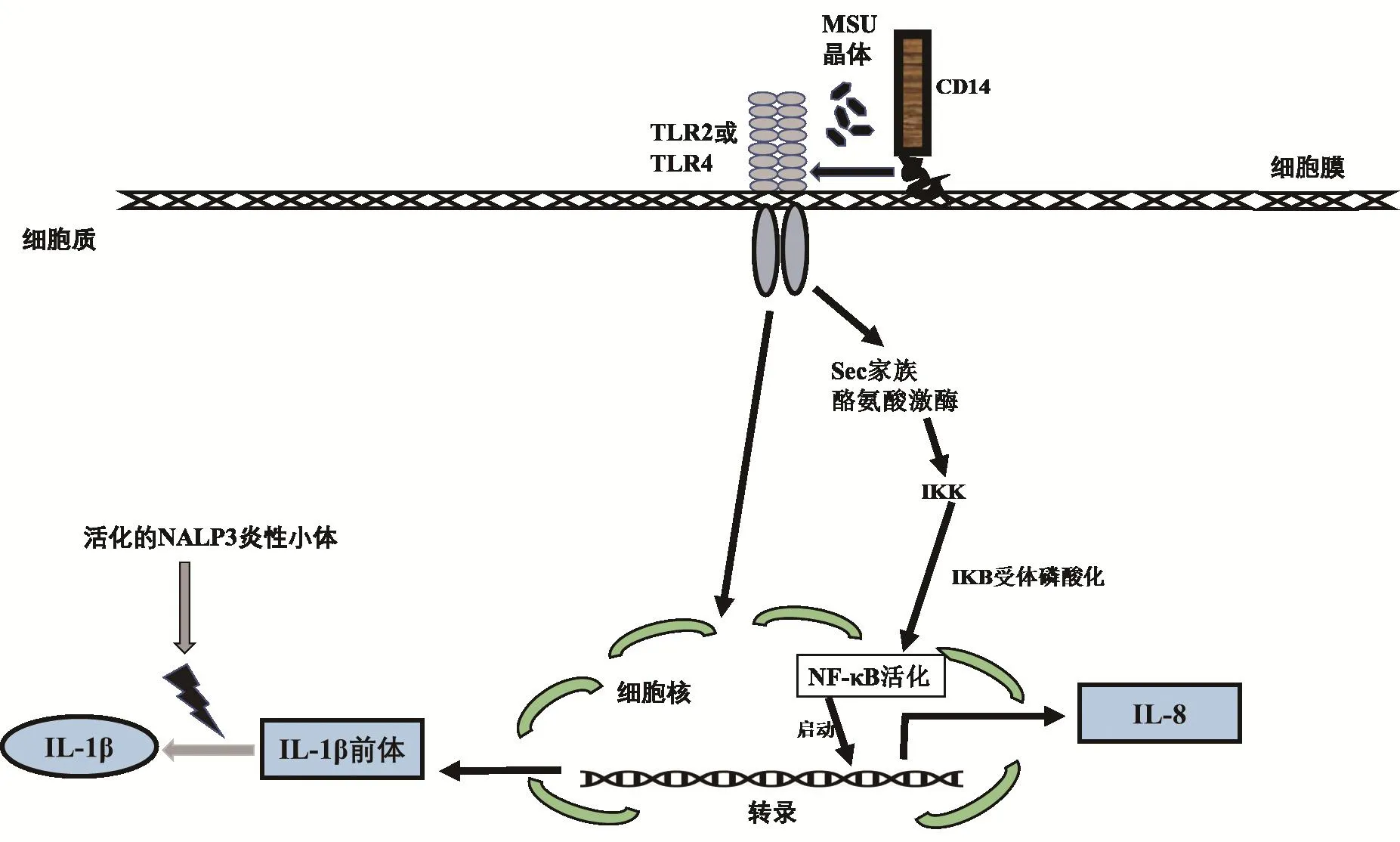
圖2 MSU結晶誘導IL-1β及IL-8表達信號轉導通路[25]
3.2 IL-8信號通路
IL-8是CXC趨化因子家族的成員,是中性粒細胞趨化因子之一,可由單核細胞、巨噬細胞、上皮細胞、內皮細胞和氣道平滑肌細胞分泌[27]。在痛風性關節炎中,大量中性粒細胞聚集在關節處形成NETs參與炎癥反應是導致發病的因素之一。體外MSU晶體誘導的單核細胞模型中,中性粒細胞大部分趨化活性來自IL-8[28]。IL-8通過促進中性粒細胞趨化,從而影響中性粒細胞在關節處的募集,影響痛風性關節炎的炎癥反應進程。在MSU晶體刺激下,Src家族酪氨酸激酶信號傳導通過刺激IKK/IκB信號通路,引發NF-κB活化誘導IL-8的表達(圖2)[23]。此外,IL-8在激活嗜中性粒細胞后,引起基質金屬蛋白酶(matrix metalloprotein, MMP)家族MMP-9和MMP-2從滑膜組織迅速釋放,兩者可在病理條件下分解與組織破壞相關的細胞外基質(extracellular matrix,ECM),進一步加重關節處的炎癥反應[29]。由此可見,IL-8在MSU晶體誘導的痛風性關節炎中發揮關鍵作用,因而抑制IL-8的釋放可能為治療該疾病提供一種新的思路。
4 小結
痛風性關節炎是由關節內和關節周圍結晶的MSU晶體引發,進而形成具有典型特征的急性炎癥。中性粒細胞NETs作為不同于壞死和凋亡的一種新型細胞死亡方式,在痛風性關節炎的免疫反應中發揮著重要作用。一方面,機體在受到MSU晶體刺激時,引發中性粒細胞在關節處大量浸潤,釋放NETs結構并促進炎癥因子分泌,進一步推動炎癥反應進程;另一方面,NETs形成有利于機體對MSU晶體的清除和炎癥消除,暫時抑制炎癥反應進程。因此,中性粒細胞NETs的形成是一把“雙刃劍”,如果形成過多而未及時清除,這種富含各種蛋白水解酶和核酸結構的膠質網狀結構能夠直接引起關節處的組織損傷,同時也可誘發無菌性炎癥反應的發生。此外,痛風性關節炎中炎癥反應的強弱、持續時間及性質受到體內各個免疫環節嚴密調控,合理地平衡體內產生和清除NETs的能力對促進關節處炎癥的自行消散過程是十分有益的。機體內的促炎過程與抗炎過程之間既相互聯系又相互制約[30],因此要合理研發既能促進自行消散又能防止炎癥進一步擴大的藥物,這為今后痛風性關節炎的臨床治療提供一個新的思路。
