頭皮上皮樣血管瘤一例
2021-05-11 12:40:22陳佳玲陳嶸祎謝嘉豪史建強
中國麻風皮膚病雜志 2021年5期
陳佳玲 陳嶸祎 謝嘉豪 史建強
1廣東醫科大學附屬醫院皮膚科,廣東湛江,524001;2南方醫科大學皮膚病醫院(廣東省皮膚病醫院),廣東廣州,510091
臨床資料患者,女,36歲。頭皮丘疹、結節3年余。3年前無名顯誘因頭皮出現綠豆至黃豆大紅色丘疹、結節,無伴瘙癢、疼痛,未予診治。皮疹增多并緩慢增大呈半球形,未見明顯融合,無破潰及出血。患者既往體健,無外傷史,無不良嗜好,家族中無類似疾病史。體格檢查:生命體征平穩,全身淺表淋巴結未觸及腫大,各系統檢查未見明顯異常。皮膚科情況:頭皮可見數個黃豆大半球形紅褐色結節,質韌,邊界清楚,互不融合,表明光滑,偶見搔抓后破潰,無糜爛滲出,無明顯壓痛(圖1a)。頭皮組織病理示:真皮內和皮下組織見大量血管腔隙組成的小葉狀團塊,境界不清,增生的血管內皮細胞肥大而圓,突向管腔,間質可見淋巴組織和大量嗜酸粒細胞浸潤(圖 2a~2c)。診斷:上皮樣血管瘤。治療:患者拒絕手術,給予得寶松(復方倍他米松)皮損內注射聯合復方丙酸氯倍他索軟膏外用,治療一周后皮疹稍消退,趨于扁平(圖1b),現已失訪。
討論上皮樣血管瘤(epithelioid hemangioma, EH)又稱血管淋巴增生伴嗜酸性粒細胞浸潤(angiolymphoid hyperplasia with eosinophilia, ALHE),是一種不常見的良性皮膚或皮下腫瘤,為各種刺激下不成熟血管和慢性炎癥細胞的局限性混合增生,炎癥細胞中常有嗜酸粒細胞[1]。表現為真皮和皮下組織的紅棕色或粉色無痛性血管結節,組織細胞樣內皮細胞增生、明顯的淋巴和嗜酸粒細胞浸潤,偶見外周血嗜酸粒細胞增多[2]。病因尚不明確,可能與皮下動靜脈分流繼發的血管畸形、創傷、感染(HTLV或皰疹病毒8)、激素分泌失調、放療及接觸砷等有關[1]。
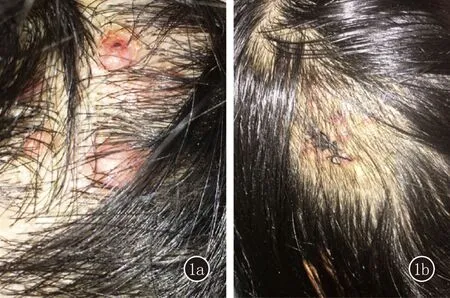
圖1 1a:治療前;1b:治療后

圖2 2a:真皮內和皮下組織見大量血管腔隙組成的小葉狀團塊,境界不清(HE,×40);2b:血管內皮細胞增生,間質見淋巴組織和嗜酸粒細胞浸潤(HE,×100);2c:增生的血管內皮細胞肥大而圓,突向管腔(HE,×400)
本病好發于 21~50歲,女性稍多見,皮損最常發生在頭頸部,其他部位例如軀干,下肢、手、陰莖、口腔黏膜和結腸很少見,常與頭相關[3]。臨床常表現為單發或多發、無痛的紅色小丘疹或結節,或斑片[4],直徑1~10 cm(0.5~3 cm常見),質韌/硬,表面光滑,可逐漸發展成紅色質硬腫物,并破潰出血[5]。病變多發時,通常成團排列或呈帶狀分布,可以融合。一般無自覺癥狀,可有輕微瘙癢或觸痛、搏動感。
在組織病理學上,上皮樣血管瘤病變表現為真皮或皮下不同大小的血管增生團塊,境界不清,血管具有肥大而圓的內皮細胞,胞漿豐富呈強嗜酸性,常顯示出空泡化和細胞質進入管腔,但無多形性及有絲分裂象,部分可見細胞實性條索。血管增生與由淋巴細胞,嗜酸粒細胞和肥大細胞組成的彌漫性炎癥浸潤混合。有時可見有生發中心的淋巴濾泡。另外,纖維化是常見的伴隨特征[6]。
結合本患者的臨床表現和病理學檢查診斷明確。
本病需與木村病(Kimura病)、上皮樣血管瘤性結節、化膿性肉芽腫、上皮樣血管內皮瘤、皮膚淋巴瘤等疾病鑒別。ALHE是血管起源的病變,木村病卻是一種慢性炎癥過程[3]。木村病好發于頜下腺、腮腺、耳前部位,常形成較大腫塊,伴淋巴結腫大,外周血嗜酸粒細胞及IgE升高,可累及腎臟等多系統自身免疫性損害,組織病理表現血管增生不明顯,無突出的管腔內皮細胞[3],可見廣泛嗜酸性微膿腫、淋巴濾泡及多核細胞。皮膚上皮樣血管瘤性結節常單發,多見于軀干及四肢,組織病理顯示腫瘤細胞及核均較大,可見含鐵血黃素沉積可沉積于瘤結節內管腔,免疫組化提示CD31、CD34 和Ⅷ因子陽性[7]。化膿性肉芽腫又稱分葉狀毛細血管瘤,常繼發于外傷后,增長迅速,不慎觸碰易出血,組織病理無明顯嗜酸粒細胞浸潤。
上皮樣血管瘤少數情況下皮損可自然消退,最有效治療為手術切除。切除可以局部復發(30%~40%),與病變下方沒有完全切除的動靜脈瘺有關。另有這些非手術常規療法,例如糖皮質激素(局部和全身應用)、甲氨蝶呤、普萘洛爾、異維A酸,冷凍療法,沙利度胺光療以及針對細胞因子的新療法,包括局部應用咪喹莫特,白介素5(mepolizumab),血管內皮生長因子A(貝伐單抗)和干擾素,激光療法也已用于治療ALHE病[8],但非手術療法復發率普遍較高。
