超高效液相色譜-串聯質譜法測定小麥粉改良劑中的苯甲羥肟酸
2021-06-04 01:43:30于文江周傳靜尹麗麗劉艷明
中國糧油學報 2021年5期
關鍵詞:方法
鄭 紅 于文江 周傳靜 尹麗麗 李 珊 劉艷明
(山東省食品藥品檢驗研究院;山東省食品藥品安全檢測工程技術研究中心,濟南 250101)
苯甲羥肟酸是一種十分重要的選礦藥劑,由于其分子的極性基團中存在著兩種帶孤對電子的氧原子和氮原子,而且這兩種原子的距離相隔較近,因此很容易與Cu2+、Ni2+、Fe3+等金屬離子形成穩定的雜環配合物[1]。但由于苯甲羥肟酸含有苯環結構及N元素,屬較難降解有機物,給人類和生態環境帶來了潛在的風險和危害[2-4]。
小麥粉是我國北方民眾最常食用的主食之一,也是許多烘焙等加工食品的主要原料來源[5-7]。近期有輿情報道小麥粉中有添加苯甲羥肟酸非食品原料的報道,添加的方式或許是通過小麥粉改良劑加入,所以建立小麥粉改良劑中苯甲羥肟酸的測定方法至關重要。小麥粉改良劑是一些天然的或化學合成的食品添加劑,根據性能及作用可分為營養強化劑類、無機鹽類、凝膠多糖類、乳化劑類、淀粉和變性淀粉類、酶制劑類、增味增色劑類、抗氧化劑類和復合添加劑類等[8-11]。綜上可得,小麥粉改良劑的種類繁多,基質較復雜,對建立小麥粉改良劑中苯甲羥肟酸的測定方法帶來較大挑戰。目前報道的測定苯甲羥肟酸的方法比較少,何旭元等[12]提出了一種采用紅外光譜溴化鉀壓片法測定苯甲羥肟酸工業品純度的方法;陳遠道等[13]報道了采用紫外光度法測定水溶液中的苯甲羥肟酸;余雪花[14]采用高氯酸鐵作顯色劑直接分光光度法測定水溶液中目標化合物的含量;周春山等[15]采用反相高效液相色譜法,對氧化鉛礦浮選藥劑銅鐵靈和苯甲羥肟酸完成了分離和測定。近期市場監督管理局針對輿情公布的補充檢驗方法BJS202002,此方法采用液相色譜法測定,前處理需氮吹濃縮、儀器運行時間耗時長,靈敏度較低,對分離度要求較高,因只有保留時間及光譜圖定性,若有陽性樣品還需質譜確證[16]。目前文獻針對苯甲羥肟酸的研究基質大多針對工業純品或水溶液,鮮見采用液相色譜-串聯質譜方法測定的報道。
1 材料與方法
1.1 材料與儀器
AB SCIEX Triple Quad6500超高效液相色譜-串聯質譜儀(配電噴霧電離源),色譜柱:Waters ACQUITY UPLC BEH C18色譜柱(75 mm×2.1 mm,1.7 μm),MS3型渦旋混合器;SB-800DTD超聲清洗儀,Sigma 3-18K型冷凍離心機,Mili-Q超純水機。
苯甲羥肟酸標準品(純度98.9%)、甲醇(色譜純)、甲酸(色譜純)和醋酸銨(色譜純)。
1.2 實驗方法
1.2.1 標準溶液配制
準確稱取苯甲羥肟酸標準品10 mg于10 mL容量瓶中,用甲醇溶解并定容至刻度線,配制成濃度為1 mg/mL的標準儲備液,于-18 ℃避光保存。準確移取苯甲羥肟酸標準儲備液,用甲醇稀釋配制成濃度為10 μg/mL的混合標準中間液,然后用甲醇∶水(1∶1)稀釋配制成濃度為5、10、20、50、100、200、500、1 000 ng/mL的系列標準工作溶液。
1.2.2 樣品前處理
準確稱取不同種類小麥粉改良劑2 g(精確至0.001 g),置于50 mL具塞離心管中,準確加入10 mL甲醇,渦旋混勻1 min,超聲提取15 min,9 000 r/min離心5 min,取上層有機層500 μL,加水500 μL稀釋一倍,渦旋混勻后過0.22 μm有機微孔濾膜過濾后,供液相色譜-串聯質譜儀測定。
1.2.3 儀器分析條件
1.2.3.1 液相色譜條件
色譜條件:色譜柱:Waters ACQUITY UPLC BEH C18色譜柱(75 mm×2.1 mm,1.7 μm);流動相A:0.1%甲酸水;流動相B:甲醇;流速:0.3 mL/min;進樣體積:5 μL;柱溫:40 ℃;洗脫方式:梯度洗脫見表1。
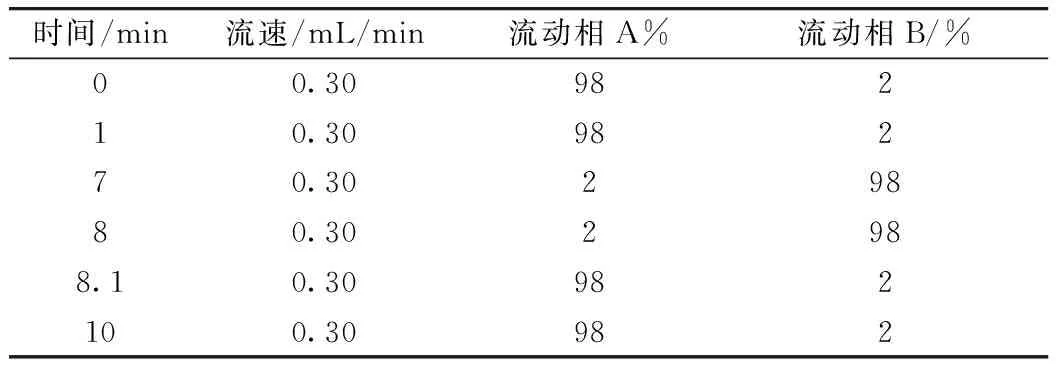
表1 超高效液相色譜-串聯質譜方法流動相梯度洗脫程序
1.2.3.2 質譜條件
離子源:電噴霧電離源負離子(ESI-);掃描方式:多反應監測;噴霧電壓:5.5 kV;霧化氣溫度:600 ℃;霧化氣壓力:55 psi;輔助氣壓力:55 psi;碰撞氣壓力:8 psi;氣簾氣壓力:35 psi。苯甲羥肟酸的質譜參數見表2。

表2 苯甲羥肟酸的質譜數、保留時間、分子式、分子量等信息
2 結果與討論
2.1 前處理條件的優化
苯甲羥肟酸為弱酸類化合物,易溶于水及甲醇。根據相似相溶的原理,實驗比較了水、甲醇/水(5∶5)、甲醇三種提取劑的回收率,對于不含淀粉或變性淀粉的小麥粉改良劑三種提取劑的回收率都在85%以上,但由于大多小麥粉改良劑中含有淀粉,前兩種提取劑由于水的存在,小麥粉改良劑呈現黏稠狀,導致樣品無法分散,實驗無法進行,只有提取劑為純甲醇時,所有樣品提取液澄清,而且回收率在90%以上,故選擇甲醇為最終提取溶劑。由于超高效液相色譜中存在溶劑化效應,當直接將甲醇提取后的樣品溶液注入UPLC-MS/MS檢測時,由于樣品溶劑(甲醇)強度遠強于初始流動相溶劑(甲醇∶0.1%甲酸水=2∶98)強度,匹配性差,導致色譜峰形分叉,當將樣品溶液用水稀釋一倍后,峰形明顯改善,而且響應值并未降低,見圖1。

圖1 苯甲羥肟酸以甲醇和甲醇/水為上機溶劑下的總離子流圖
2.2 液相色譜質譜條件優化
本研究采用蠕動泵直接注入苯甲羥肟酸標準溶液(1 μg/mL)優化各參數。苯甲羥肟酸分子結構見圖2,分子量為137,在Q1全掃描模式下失去氫生成母離子[M-H]-;然后采用Product ion 模式,通過改變碰撞能,對子離子進行掃描,圖2為碰撞能30 eV下,苯甲羥肟酸的子離子掃描圖,可看出苯甲羥肟酸碎片信息很豐富,主要為121、92、77、65、58的碎片離子。然后將Q1 MS模式下找到的母離子[M-H]-及Product ion 模式找到的所有碎片離子導入MRM掃描模式的列表中,對碰撞能等參數進行系統優化,發現所有碎片離子中92和58的豐度最高,而且在實際樣品的測定中信號穩定且無干擾色譜峰存在,因此本實驗最后選以上兩個碎片作為最終的監測離子。

圖2 苯甲羥肟酸的子離子掃描譜圖(30 eV)
此外比較甲醇-水、甲醇-醋酸銨、甲醇-甲酸水三種流動相系統對其峰形及響應值考察,結果顯示三種流動相體系下,靈敏度差異不大,但當采用甲醇-甲酸水為流動相時,苯甲羥肟酸峰形最佳,故選擇甲醇-甲酸水為最終流動相體系。
2.3 基質效應
超高效液相色譜-串聯質譜方法雖然具有液相優異的分離度和質譜較高的分辨力,但基質效應的存在嚴重影響了定量的準確性和精密度。為了考察不同種類小麥粉改良劑的基質效應,本研究采用比較陰性基質標準工作溶液和純溶劑標準工作溶液的擬合標準曲線的斜率,進而評估小麥粉改良劑的基質效應。以公式A=Ks/Kp計算基質效應,其中Ks為含基質標準工作曲線的斜率,Kp為純溶劑標準工作曲線的斜率,A為兩斜率之比,當A≈ 1時,基質效應為0;A>1,說明有基質效應增強,A<1 說明基質效應抑制。表3為濃度為5~1 000 ng/mL線性范圍內,純溶劑及不同陰性基質匹配溶液校正曲線下的標準曲線方程、斜率及A值,發現4類小麥粉改良劑(無機鹽類、淀粉和變性淀粉類、酶制劑類和復合添加劑類)的A值在0.980~1.037之間,偏差在-2%~3.7%,A≈1,基質效應可忽略,表明本方法去除基質效應能力強,免除了每類小麥粉改良劑需配制一套基質匹配校正曲線的繁瑣操作。
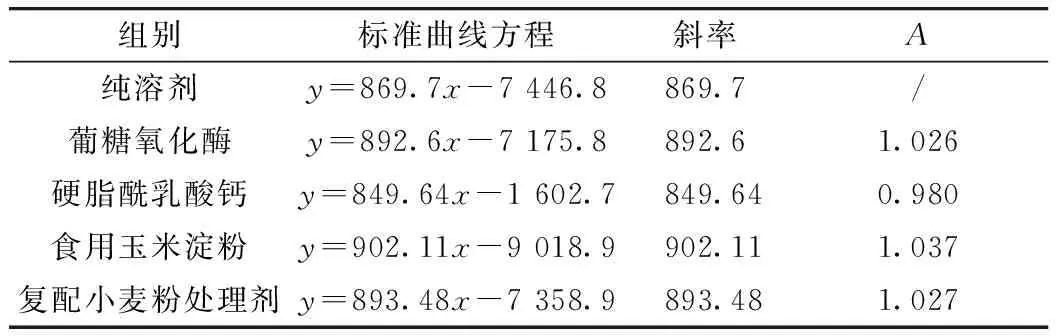
表3 不同空白基質下苯甲羥肟酸的標準曲線方程、斜率及A值
2.4 方法的線性范圍、檢出限與定量限
將1.2.1配制的標準工作溶液分別注入超高效液相色譜-串聯質譜儀檢測,以標準工作溶液峰面積為縱坐標,以標準溶液的濃度為橫坐標,權重為1/X時,繪制標準曲線,結果表明HPLC- MS/MS方法在濃度為5~1 000 ng/mL范圍內,r2大于0.999,線性良好。
采用在空白小麥粉改良劑中添加目標化合物的方法,確定方法的檢出限與定量限。以對應色譜峰響應值3倍信噪比的質量濃度作為方法檢出限,對應色譜峰響應值10倍信噪比的質量濃度作為方法定量限,見表4。
2.5 方法回收率和精密度
采用在陰性小麥粉改良劑中添加定量限、2倍定量限、10倍定量限的目標化合物進行加標回收試驗,驗證方法準確度和精密度。每個濃度水平平行測定6次取平均值,計算回收率和精密度。結果見表4。陰性樣品及添加水平為定量限的MRM色譜圖見圖3。

圖3 陰性小麥粉改良劑的MRM譜圖(a,b)和添加量為定量限(0.0 5 mg/kg)的小麥粉改良劑的MRM譜圖(c,d)

表4 苯甲羥肟酸在UPLC-MS/MS上的檢出限、定量限、線性范圍、加標回收率和精密度
2.6 與標準方法比較
將本方法與BJS202002比較,見表5。發現此方法的檢出限不僅比標準BJS202003降低了10倍,而且省去了氮吹濃縮的時間,減少了儀器運行的時間,大大提高了檢測效率。此外質譜定性比紫外光譜定性更準確。
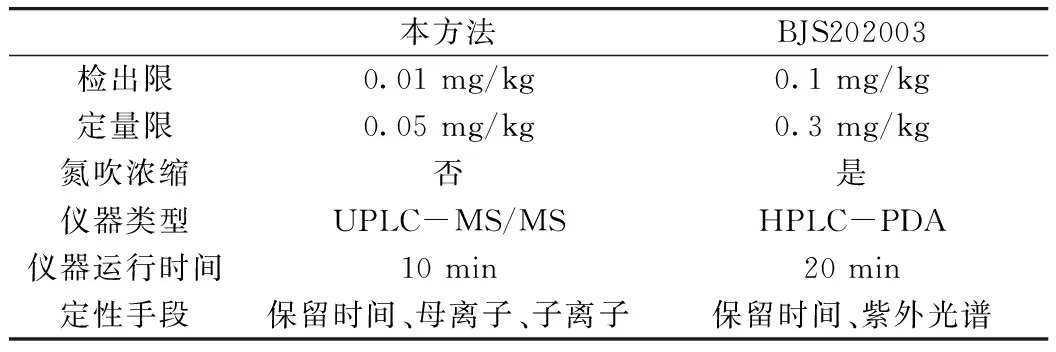
表5 本方法與BJS202003比較
2.7 樣品測定
對市售的28個小麥粉改良劑的苯甲羥肟酸采用本方法進行測定,方法適用性好、回收率高。其中有一批次有檢出,檢出值為1.59 mg/kg,其譜圖見圖4,其他均未發現有陽性樣品檢出。結果表明,小麥粉改良劑中確實存在苯甲羥肟酸檢出,應當引起相關監管部門及生產企業的重視,完善相關的檢測及限量標準。
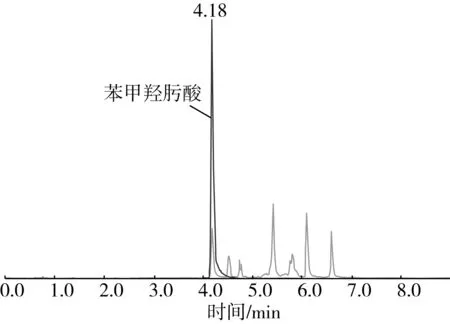
圖4 陽性小麥粉改良劑的總離子流圖
3 結論
本研究系統考察了不同提取劑對小麥粉改良劑中苯甲羥肟酸的提取效果,優化了色譜條件及質譜參數,并考察了不同種類小麥粉改良劑的基質效應,建立了小麥粉改良劑中苯甲羥肟酸的超高效液相色譜-串聯質譜方法。該方法與BJS202002比較,簡單、省時、靈敏度高、特異性強。此外基質效應可忽略,適用于大批量及種類繁多的小麥粉改良劑中苯甲羥肟酸的測定,提高了檢驗工作的工作效率;同時為建立相關標準及監管部門風險監控提供參考。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
