超高效液相色譜-串聯質譜法測定醬鹵肉制品中1-甲基咪唑、2-甲基咪唑及4-甲基咪唑
2021-06-18 01:01:30閔宇航黃璐瑤余曉琴
食品工業科技 2021年10期
關鍵詞:實驗
閔宇航,黃璐瑤,余曉琴,王 穎
(四川省食品藥品檢驗檢測院,四川成都 610097)
焦糖色是一種食品添加劑,不僅能起到增色增香的作用,還有助于提高食品品質,被廣泛應用于醬油、食醋、料酒、烘焙食品、糖果、飲料等多種食品的生產中。焦糖色按生產工藝可以分為四類:Ⅰ類為普通法焦糖色,Ⅱ類為苛性亞硫酸鹽法焦糖色,Ⅲ類為氨法焦糖色,Ⅳ類為亞硫酸銨法焦糖色。在焦糖色的生產過程中主要發生了美拉德反應和焦糖化反應。普通的焦糖色主要發生焦糖化反應,而加入了氨(銨)作為催化劑的Ⅲ、Ⅳ類焦糖色以美拉德反應為主。與非氨(銨)法生產相比,氨(銨)法生產焦糖色具有反應快、周期短、呈色深的特點,但是會產生美拉德反應的伴生危害產物2-甲基咪唑和4-甲基咪唑。
4-甲基咪唑具有較強的驚厥作用,可以使動物產生超興奮狀態從而發生痙攣[1]。2007年美國國家毒理學規劃處(National Toxicology Program,NTP)將4-甲基咪唑作為可致癌物質發出警告[2],美國加州環境健康危害評估辦公室(OEHHA)2011年將4-甲基咪唑列入致癌化學品清單(the Proposition 65 list)中,并設定4-甲基咪唑的致癌潛能為0.024 (mg/kg bwday),無顯著風險水平(NSRL)為29 μg/d,要求超過該安全值的食品必須帶有警告性標識[3]。2013年,世界癌癥研究機構(IARC)致癌物分級目錄將4-甲基咪唑、2-甲基咪唑列入2B級致癌物[4]。1-甲基咪唑與4-甲基咪唑、2-甲基咪唑互為同分異構體,其毒理學性質沒有得到充分的調查,但在部分食品中發現1-甲基咪唑的存在[5]。醬鹵肉制品是日常消費量較大的食品,但是目前并沒有醬鹵肉制品中甲基咪唑類化合物含量的研究報道。GB 2760-2014中規定4類焦糖色均不能在醬鹵肉制品中使用[6],醬鹵肉制品中的甲基咪唑類污染物可能來源于生產者非法添加了Ⅲ、Ⅳ類焦糖色,也可能來源于配料中醬油、醋等調味品的帶入。
目前報道食品中檢測的甲基咪唑類化合物多為2-甲基咪唑和4-甲基咪唑,缺少1-甲基咪唑的檢測。檢測方法有分光光度法[7-8]、拉曼光譜法[9]、氣相色譜法[10-12]、氣相色譜質譜聯用法[13-15]、高效液相色譜法[16-17]和液相色譜質譜聯用法[18-20]。分光光度法和氣相色譜法的前處理較為繁瑣[21-22];拉曼光譜儀普及度較低;高效液相色譜法的靈敏度較低。本實驗期望結合超高效液相色譜的快速分離能力以及質譜的高選擇性、高靈敏度,建立一種快速、準確、穩定的UPLC-MS/MS法測定醬鹵肉制品中的1-甲基咪唑、2甲基咪唑和4-甲基咪唑,填補醬鹵肉制品中甲基咪唑類化合物檢測的空白,為以后監管部門的風險評估和限值制定打下了基礎。
1 材料與方法
1.1 材料與儀器
Waters Oasis MCX固相萃取小柱(150 mg,6 mL) 美國Waters公司;1290 Infinity Ⅱ超高效液相色譜儀、 6460C三重四極桿質譜儀 美國Agilent公司;Thermo Scientific HeraeusMlutifuge X3R高速冷凍離心機 美國Thermo Fisher Scientific公司;IRM IDH30超聲儀 德國IRM Technology GmbH公司;IKA MS3渦旋混合器 德國IKA公司;BiotageTurboVap全自動氮吹儀 瑞典Biotage公司;HM100 POWTEQ刀式研磨儀 北京格瑞德曼公司。
1.2 實驗方法
1.2.1 標準溶液的制備 準確稱取1-MEI、4-MEI標準品10 mg(精確至0.01 mg),用乙腈溶解并定容至10 mL,混勻,制成1 mg/mL的標準儲備液。精密吸取10 μL 1-MEI、4-MEI標準儲備液以及100 μL 2-MEI的標準品置10 mL容量瓶中,乙腈定容至刻度,混勻,制成1 μg/mL的混合標準溶液。
準確稱取1-MEI-D6、2-MEI-D6、4-MEI- D6標準品10 mg,用乙腈溶解并定容至10 mL,混勻,制成1 mg/mL的內標儲備液。再精密吸取10 μL 1-MEI-D6、2-MEI- D6、4-MEI- D6的內標儲備液置10 mL容量瓶中,乙腈定容至刻度,混勻,制成1 μg/mL的混合內標溶液。
精密吸取混合標準溶液和混標內標溶液適量,用初始流動相稀釋成30、60、100、150、200、300、400 ng/mL混合系列標準工作溶液,其中1-MEI-D6、2-MEI- D6、4-MEI- D6的濃度均為50 ng/mL。
1.2.2 供試品溶液的制備 取樣品約200 g,用刀式研磨儀充分均質,裝入潔凈的容器中。
稱取上述均質后的樣品2 g(精確至0.001 g),置50 mL離心管中,加入混合內標溶液100 μL,加水20 mL,渦旋1 min,再超聲(功率500 W)5 min,以10000 r/min在4 ℃條件下離心5 min,上清液待凈化。
凈化前,依次用5 mL甲醇,5 mL水對MCX固相萃取小柱進行活化。再精密量取10 mL上清液,過MCX固相萃取小柱,依次用2%甲酸水溶液5 mL,甲醇5 mL淋洗,再用5%氨水甲醇溶液10 mL洗脫,收集全部洗脫液,45 ℃氮吹至近干,精密加入初始流動相1 mL復溶,過0.22 μm有機系濾膜,濾液供液相色譜-串聯質譜儀測定。
1.2.3 分析條件 液相色譜條件:色譜柱:Agilent Zorbax HILIC色譜柱(2.1 mm×100 mm,3.5 μm);柱溫:35 ℃;進樣量:5 μL;流動相:A相:乙腈;B相:5 mmol/L乙酸銨溶液;流速0.6 mL/min;流動相梯度見表1。
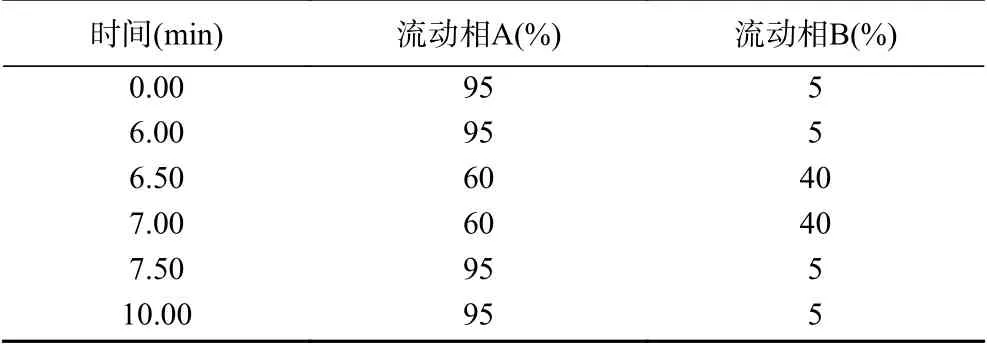
表1 流動相梯度洗脫表Table 1 Gradient elution program
質譜條件:離子源:電噴霧離子源(ESI);掃描方式:正離子掃描;檢測方式:多反應檢測(MRM);毛細管電壓:3.5 kV;干燥氣溫度:325 ℃;干燥氣流量:11 L/min;霧化氣壓力:35 psi;鞘氣溫度:325 ℃;鞘氣流量:11 L/min。化合物質譜參數見表2。
“知識改變命運”,這是現代人公認的真理。可惜的是,在懵懂的轉型時期,經受著陣痛的人們對此并沒有清晰的認知。在此情況下,他們既無法與伴侶進行良好的溝通,更難以擺脫貧困潦倒的生存現實。因為不明就里,即便有偶而的掙脫,也顯得蒼白無力。
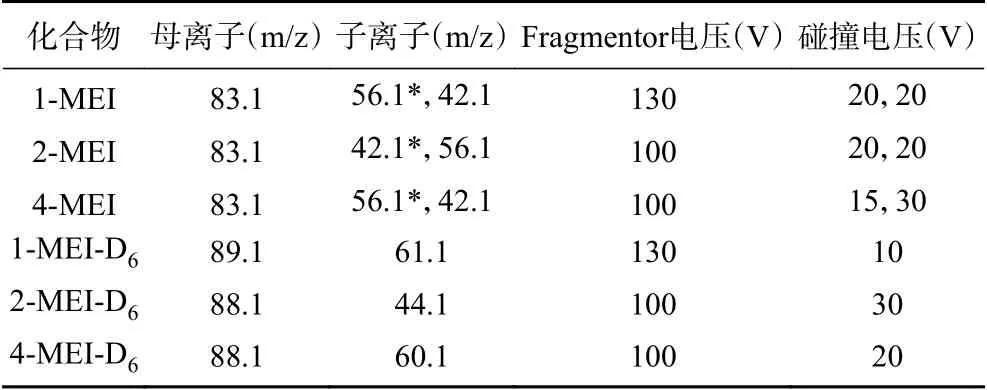
表2 1-MEI、2-MEI、4-MEI、1-MEI-D6、2-MEI-D6、4-MEI-D6的質譜參數Table 2 Mass parameters of 1-MEI, 2-MEI, 4-MEI,1-MEI-D6, 2-MEI-D6, 4-MEI-D6
1.3 數據處理
采用美國Agilent Technologies Inc.的MassHunter Workstation Software 06.00軟件進行分析處理,Microsoft Office Excel軟件進行統計分析。
2 結果與分析
2.1 色譜條件的優化
2.1.1 色譜柱的選擇 1-MEI、2-MEI、4-MEI這3種化合物互為同分異構體,具有相同的分子質量和離子碎片,需要通過色譜柱實現完全分離后才能進行檢測[23]。由于目標化合物為含氮極性小分子化合物,采用常用的十八烷基硅烷鍵合硅膠柱可能會出現保留差、出峰快、分離效果不佳的情況[24]。實驗首先嘗試了Merck Purospher STAR LP RP-18 endcapped(4.6 mm×250 mm,5 μm),3種化合物在填料為C18的色譜柱上無法達到很好的分離效果。實驗又選擇了適合高極性化合物的HILIC填料的色譜柱進行分離。分別比較了Thermo Acclaim Mix-Mode HILIC-1(2.1 mm×150 mm,3 μm)、Agilent Zorbax HILIC Plus(2.1 mm×100 mm,3.5 μm)、Agilent Poroshell 120 HILIC(2.1mm×100 mm,2.7 μm)、Waters ACQUITY UPLC BEH HILIC(2.1 mm×100 mm,1.7 μm)4種不同品牌和參數的HILIC色譜柱。最終選擇了Agilent Zorbax HILIC(2.1 mm×100 mm,3.5 μm),可以實現目標化合物的很好分離(見圖1)。
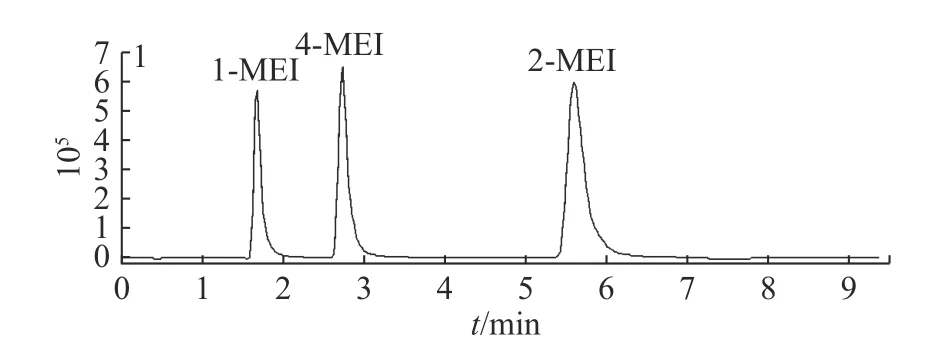
圖1 醬鹵肉基質中1-MEI, 2-MEI和4-MEI的色譜圖Fig.1 Chromatogram of 1-MEI, 2-MEI and 4-MEI in sauced meat products
2.1.2 流動相的優化 乙腈是HILIC色譜柱常用的有機流動相,而在流動相水相中加入緩沖鹽有利于提高色譜柱的柱效和重復性[25-26]。實驗比較了不同梯度條件的乙腈-5 mmol/L的乙酸銨溶液。在表1的梯度條件下,目標化合物可以達到一個很好的分離。
2.2 質譜條件的優化
分別取用1-MEI、2-MEI、4-MEI、1-MEI-D6、2-MEI-D6、4-MEI-D6標準溶液,直接進樣進行質譜條件優化。在ESI +模式下分別進行全掃描,確定1-MEI、2-MEI、4-MEI的母離子均為m/z 83.1,對其母離子進行二級質譜子離子掃描,3種待測化合物均分別丟失-C2H3和-NC2H3,相應產生碎片離子m/z 56.1和m/z 42.1;在ESI+模式下分別進行全掃描,1-MEI-D6、2-MEI-D6、4-MEI-D6均出現m/z 89.1和m/z 88.1兩個母離子峰,1-MEI-D6的m/z 89.1響應高于m/z 88.1,實驗選擇m/z 89.1作為1-MEID6的母離子;而2-MEI-D6和4-MEI-D6的m/z 88.1響應高于m/z 89.1,實驗選擇m/z 88.1作為2-MEI-D6和4-MEI-D6的母離子。對三種內標化合物的母離子進行二級質譜子離子掃描,分別產生碎片離子m/z 61.1、m/z 60.1、m/z 44.1,作為內標化合物的定量離子。
2.3 前處理條件的優化
2.3.1 提取溶劑的選擇 根據3種甲基咪唑類化合物物理和化學性質,實驗采用添加水平為50 μg/kg的基質加標實驗,每個考察條件做6個平行實驗(n=6)。考察水、甲醇、乙腈3種提取溶劑的提取能力,以加標回收率考察提取效果(見圖2)。結果表明,1-MEI:水>乙腈>甲醇;4-MEI:水>甲醇>乙腈;2-MEI:水與甲醇效果相似,均明顯優于乙腈。綜上,同時考慮成本、環保等因素,選擇水為提取溶劑。
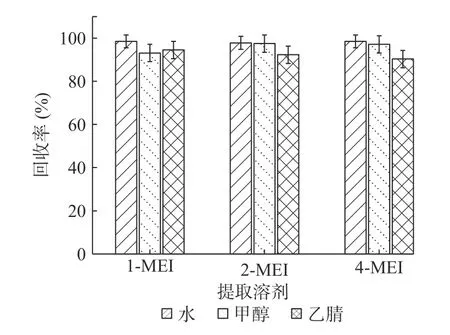
圖2 提取溶劑的考察(n=6)Fig.2 Research of extraction solvents(n=6)
2.3.2 固相萃取小柱的選擇 甲基咪唑類化合物由于其分子中含有氮原子,凈化時宜采用對堿性物質有較好保留的陽離子交換固相萃取柱[27]。因此實驗比較了市面上常見的兩種品牌的陽離子交換固相萃取小柱,分別為Waters Oasis MCX(150 mg,6 mL)和Agela Cleanert PCX(150 mg,6 mL)。采用添加水平為50 μg/kg的基質加標實驗(n=6),以加標回收率考察兩個小柱的凈化效果(見圖3)。結果表明Waters Oasis MCX凈化效果略優于Agela Cleanert PCX。
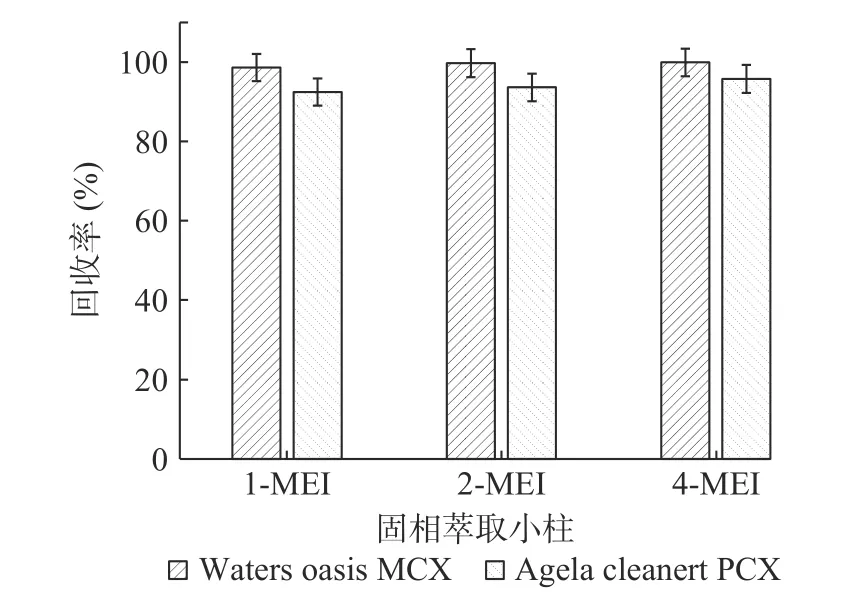
圖3 固相萃取小柱的考察(n=6)Fig.3 Research of solid phase extraction colum(n=6)
2.3.3 固相萃取條件的優化 實驗采用添加水平為50 μg/kg的基質加標實驗(n=6),以加標回收率考察了不同濃度的氨水-甲醇溶液對目標化合物的洗脫能力,分別考察了2%、5%、10%、20%的氨水-甲醇溶液(見圖4)。四種溶液對2-MEI、4-MEI的洗脫效果無明顯差異,其中1-MEI洗脫受氨水濃度的影響稍大,從實驗結果來看,選擇5%氨水-甲醇溶液作為洗脫溶劑。
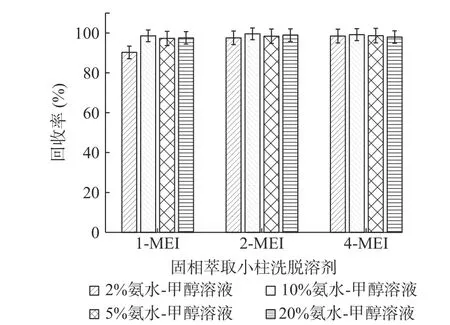
圖4 MCX固相萃取小柱洗脫溶劑的考察(n=6)Fig.4 Research of the elution solvent on MCX solid phase extraction column(n=6)
實驗考察了不同體積的洗脫溶劑的對目標化合物的洗脫能力。分別考察了5、10、15 mL的洗脫溶劑用量,以加標回收率考察不同用量洗脫溶劑的洗脫效果(見圖5)。結果發現使用10 mL及15 mL洗脫溶劑的洗脫效果接近,均優于5 mL的洗脫效果。綜合洗脫效果、成本及環保等因素,選擇10 mL作為洗脫體積。
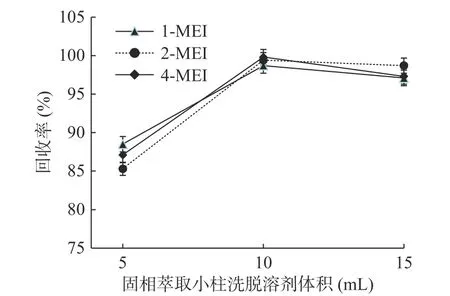
圖5 MCX固相萃取小柱洗脫溶劑體積的考察(n=6)Fig.5 Research of the volume of the elution solvent(n=6)
2.4 方法學驗證
2.4.1 基質效應 實驗通過提取后添加法來評價基質效應(matrix effect, ME),將空白樣品按前處理方法進行提取凈化后,在空白樣品提取液中添加目標化合物,用已定的色譜、質譜條件進行檢測,然后與同樣濃度的純溶劑中目標化合物的離子響應強度進行比較。采用公式:其中A和B分別表示純溶劑與基質溶液中分析物的離子響應強度。若ME<1,說明基質對分析物的響應產生了抑制作用;ME>1,說明基質會增強分析物的響應;ME=1,說明不存在基質效應。實驗考察了三個濃度水平的基質效應(30、200、400 ng/mL),1-MEI三個濃度水平下的ME分別為0.65、0.55、0.57;2-MEI三個濃度水平下的的ME分別為0.78、0.74、0.77;4-MEI三個濃度水平下的的ME分別為0.54、0.57、0.58.結果說明基質對3種咪唑類化合物均有程度不一的抑制作用。實驗采用同位素內標法定量以消除基質效應的影響,保證定量的準確性[28-29]。
2.4.2 線性范圍、檢出限和定量限 用初始流動相配制1-MEI、2-MEI和4-MEI的混合系列標準工作溶液,濃度分別為30、60、100、150、200、300、400 ng/mL。以目標化合物定量離子的峰面積與相應內標化合物定量離子的峰面積的比值為縱坐標,質量濃度為橫坐標得到線性回歸方程(見表3),3種甲基咪唑化合物線性相關系數均大于0.9997。通過空白樣品加標測定,3種甲基咪唑類化合物在10 μg/kg的添加水平下經過前處理所得試液的定性離子信噪比(S/N)均大于3,表明該方法的3種甲基咪唑類化合物的檢出限均可達到10 μg/kg;3種甲基咪唑類化合物在30 μg/kg的添加水平下經過前處理所得試液的定性離子信噪比(S/N)均大于10,且1-MEI、2-MEI和4-MEI的回收率分別為106.0%、100.6%、92.0%,表明該方法3種化合物的定量限均可達到30 μg/kg。
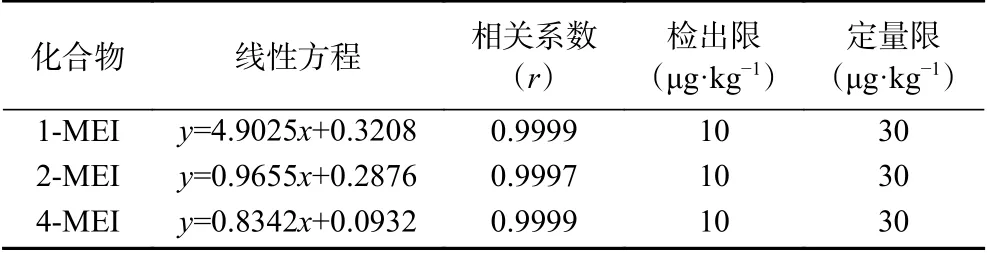
表3 1-MEI、2-MEI和4-MEI的線性方程、相關系數、檢出限及定量限Table 3 Regression equation, correlation coefficient, limits of detection (LOD) and limits of quantitation(LOQ) of 1-MEI, 2-MEI and 4-MEI
2.4.3 回收率、精密度和穩定性 選取醬鹵肉樣品中添加30、60、300 μg/kg三個水平,每個水平分別做6份平行樣,計算回收率和相對標準偏差(RSD)。3種甲基咪唑類化合物三個加標水平下的平均回收率為92.0%~116.3%,RSD為0.70%~3.96%。樣品加標溶液分別在0、2、4、8、16、24 h進樣,3種甲基咪唑類化合物的定量離子峰面積RSD為2.69%~4.41%。結果表明該方法的準確、穩定、可靠。
2.4.4 實驗室間協作驗證 為驗證本方法的有效性和適用性,選擇了6家檢測機構對本方法進行驗證。6家檢測機構的結果顯示,3種甲基咪唑類化合物在30~400 ng/mL范圍內線性關系良好,相關系數(r)均大于0.9990;方法檢出限濃度下的定性離子信噪比均大于3;方法定量限濃度下的定性離子信噪比均大于10。30、60、300 μg/kg三個加標水平的平均回收率為77.2%~115.2%,RSD為0.80%~9.34%。
2.5 實際樣品的檢測
采用本方法對市售的36批次醬鹵肉樣品進行測定。結果發現,36批次樣品均檢出4-MEI,含量為3.1~306.6 μg/kg;2批次樣品檢出2-MEI,含量為21.4~23.6 μg/kg;36批次樣品均未檢出1-MEI。從結果分析,醬鹵肉制品中可能存在超范圍使用Ⅲ、Ⅳ類焦糖色的情況,也可能是由其配料帶入,例如,醬油、醋等,需要結合其生產工藝進行綜合分析。
3 結論
本實驗建立了醬鹵肉制品中3種甲基咪唑類化合物測定的超高效液相色譜串聯質譜方法,樣品經水提取,MCX固相萃取柱凈化后上機檢測。方法操作簡便、靈敏度高、結果準確可靠,能一次性測定醬鹵肉制品中3種甲基咪唑類化合物,可以滿足監管需要,為以后醬鹵肉制品中甲基咪唑類化合物分析測定以及相關國家標準的制定提供了重要參考價值。
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
