超快速固相萃取-氣相色譜-質譜法測定水中49種半揮發性有機物
2021-06-30 06:01:34陳興定姚志建
環境科技 2021年3期
關鍵詞:檢測
焦 偉,耿 旭,李 樂,陳興定,姚志建
(1.南京大學環境學院,江蘇 南京 211046,2.江蘇國創環保科技有限公司,江蘇 南京 211106)
0 引言
隨著我國經濟的快速發展,大量工業廢棄物、殘留農藥、生活垃圾及污水進入水體,導致水體有機物污染,成為當前人們重點關注的環境污染問題之一。半揮發性有機物(SVOCs)是多環芳烴類、氯苯類、硝基苯類和鄰苯二甲酸酯類等化合物的統稱,大部分為內分泌干擾物(EDCs),具有致畸、致癌和致突變的“三致”毒性作用,多種SVOCs 被我國生態環境部和美國環保部(EPA)列為優先監測的有機污染物“黑名單”[1-2]。
水中SVOCs 含量少、種類復雜,各組分的理化性質相差較大[3],傳統的檢驗方法多采用氣相色譜法,需要采用不同的有機溶劑進行樣品前處理,然后選用電子捕獲檢測器(ECD)、火焰光度檢測器(FPD)等不同的檢測器分別測定,方法不統一且不具備定性確證能力。而固相萃取(solid phase extraction,SPE)作為一種新型的樣品前處理技術已廣泛應用于水中痕量有機污染物的富集和濃縮,在很大程度上彌補了傳統預處理方法的不足,但是傳統的固相萃取過程耗時長,固相萃取小柱的填料直接影響檢測靈敏度。固相萃取與氣相色譜-質譜聯用技術相結合能更好地分離和檢測有機物[4-7];本研究采用課題組自主開發的具有親疏水結構的多孔吸附材料作為固相萃取劑[8],利用蠕動泵、聚四氟乙烯管路、壓力/流量控制系統等零部件設計了一種大體積快速固相萃取系統,以多環芳烴、增塑劑、有機氯和有機磷農藥等49種SVOCs 為研究目標,參考美國EPA 的檢測方法,在GB/T 5750.8—2006《生活飲用水標準檢驗方法有機物指標》附錄B 的基礎上,對儀器性能、內標回收率等進行了嚴格的質量控制和質量保證,對固相萃取條件等進行方法學研究與探討,建立飲用水中多種SVOCs 快速的前處理技術和檢測方法。
1 實驗部分
1.1 儀器、試劑與標準溶液
儀器:氣相色譜-質譜聯用儀(Trace1300+ISQ7000、EI 電離源、毛細管色譜柱:TG-5SILMS、毛細管柱:30 m × 0.25 mm × 0.25 μm);旋轉蒸發儀(IKA RV8);自動定量濃縮儀(MuLtiVap-10);固相萃取柱和快速固相萃取系統均為自制。
試劑:甲醇(CH3OH,分析純)、二氯甲烷(分析純)、乙酸乙酯(分析純)、氫氧化鈉(NaOH,分析純),均購自國藥集團化學試劑有限公司。
標準溶液:49 種半揮發性有機物混合標準使用液(質量濃度為200 μg/mL);替代物(1,3-二甲基-2-硝基苯、苝-d12、磷酸三苯酯,質量濃度為500 μg/mL);內標物(苊-d10、菲-d10、艸屈-d12,質量濃度為1 000 μg/mL),均購自上海安譜公司。
1.2 配置標準曲線
分別吸取質量濃度為200 μg/mL 的混合標準溶液2.5,5,10,25,50 μL,加入質量濃度為10 μg/mL的內標溶液40 μL,加入質量濃度為10 μg/mL 的替代物溶液40 μL,用二氯甲烷準確定容至1.0 mL,混勻,含內標和替代物質量濃度均為0.40 μg/mL,配成不同濃度梯度的混合標準工作溶液。吸取1.0 μL 標準工作溶液進行測定,分別以目標離子與內標物的峰面積比值對目標離子的濃度作圖,繪制標準曲線。
1.3 超快速固相萃取過程
超快速固相萃取系統由液體供給部分和萃取/洗脫轉換部分組成。液體供給由2 臺蠕動泵組成,可以實現待測液體進樣、污染物洗脫以及整個管路清洗功能;萃取/洗脫轉換部分由六通閥構成,能實現對待測物萃取和洗脫的即時切換。萃取流程見圖1。
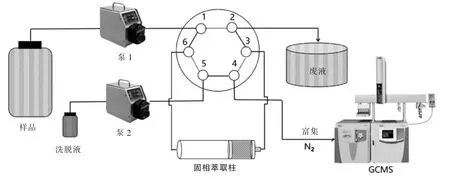
圖1 快速固相萃取流程示意
進樣模式:將500 mg 固相萃取劑填裝于固相萃取柱中,此時六通閥內的液體流路如圖1 實線所示,調整蠕動泵1 的流速,使待富集樣品以一定流速通過固相萃取柱,開啟富集過程。
洗脫模式:將六通閥轉換至洗脫模式,調整蠕動泵2 的流速,使得洗脫液以一定流速通過固相萃取柱,開啟洗脫過程。用玻璃試管接收洗脫液,過濾、氮吹濃縮、定容后即可上機檢測。
1.4 色譜條件
GCMS 分析采用的色譜柱進樣口溫度為280 ℃。程序升溫過程:35 ℃恒溫保持2 min,15 ℃/min 升溫至150 ℃保持5 min,以3 ℃/min 的速度升溫至290 ℃,保持2 min。EI 離子源溫度為280 ℃,傳輸線溫度為300 ℃。
2 結果與分析
2.1 儀器條件選擇
49 種SVOCs 使用TG-5SILMS 毛細管柱完全可以滿足分離的要求。用SCAN 方式對目標化合物進行分析得到質譜圖,選擇具有結構特征的碎片,并考察其豐度比得到相應的特征離子,各目標化合物的保留時間、特征離子和參比化合物見表1。
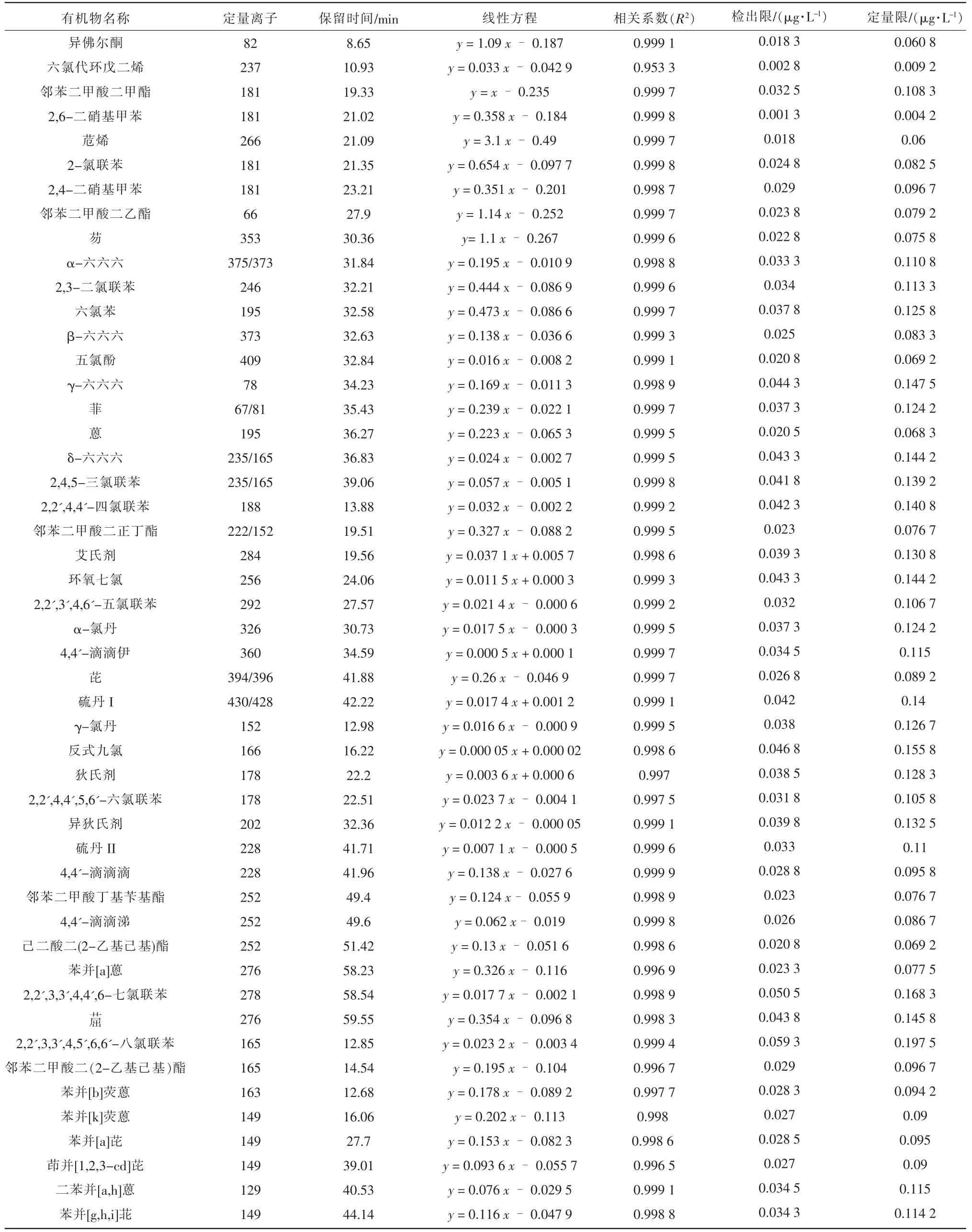
表1 49 種SVOCs 的定量離子、保留時間、線性方程、檢出限、定量限
2.2 固相萃取柱的選擇及流速控制
通過對C18 固相萃取柱與本課題組自制的兩親固相萃取柱的富集效果進行比較,發現C18 柱對強極性化合物如毒死蜱與對硫磷、七氯與馬拉硫磷、莠去津與五氯酚這3 組化合物的萃取效率較差,回收率只有30%~40%。
課題組研發的新型兩親材料不但吸附容量大,而且吸附平衡時間極短。普通的固相萃取裝置的流速只能控制在20 mL/min 以內,富集2 L 水樣需要100 min,消耗時間長。本實驗自制了正壓型快速固相萃取裝置,萃取流速最快可達到1 L/min,研究使用自制兩親固相萃取柱和自制快速固相萃取裝置對比了5,10,20,100,200,300 mL/min 的流速對富集效率的影響,最終發現,萃取流速在200 mL/min 以內時,萃取效率沒有明顯變化。
2.3 有機改進劑的選擇
有機改進劑的添加會使柱填料鍵合相表面含有一定濃度的有機溶劑薄層,從而改善強憎水組分在柱填料表面的吸附能力,同時,有機改進劑還可以增加非極性有機物在水溶液中的溶解度,減少其在容器壁上的吸附,能夠大幅度提高物質的回收率。
使用低毒的甲醇[9-10]作為改進劑,在裝有2 L 空白水樣的樣品瓶中分別加入5,10 和15 mL 甲醇,再添加中濃度加標水平的標準溶液,比較改進劑加入量對SVOCs 回收率的影響,結果顯示:改進劑添加量從5 mL 增加至10mL 時,六氯苯、六六六、滴滴涕等化合物的回收率從60%提升至70%左右;改進劑添加量從10 mL 增加至15 mL 時,回收率沒有明顯區別。因此確定改進劑加入量為10 mL。
2.4 洗脫劑的選擇
有學者對比了正己烷、丙酮、甲醇、二氯甲烷和乙酸乙酯對已富集SVOCs 的色譜柱的洗脫效果,結果發現乙酸乙酯-二氯甲烷混合溶液洗脫效果最好[11]。本研究比較了洗脫液中乙酸乙酯和二氯甲烷的比例,結果表明∶采用乙酸乙酯-二氯甲烷混合溶液(體積比為1 ∶2)時效果最好,尤其對多環芳烴組分的回收率影響最大。
2.5 方法學評價與質量控制
為了消除干擾以準確反映目標化合物與響應值間的對應關系,本試驗采用內標法,用標準工作曲線中目標化合物定量離子的峰面積與相應的內標物定量離子的峰面積之比對目標化合物的濃度進行線性回歸。結果表明:49 種SVOCs 線性關系良好,R2均大于0.995,其中異佛爾酮、六氯代環戊二烯、鄰苯二甲酸二甲酯等30 種化合物的R2均大于0.999。最低濃度混合標準工作溶液跨時4 d 檢測,按3 和10 倍信噪比計算方法最低檢出限和定量限,2 L 水樣富集后洗脫液濃縮至1 mL 進行檢測,方法檢出限為0.001~0.059 μg/L,方法定量限為0.004~0.198 μg/L,標準曲線范圍為0.5~10 mg/L。
分別在2 L 純水中添加低、中、高3 種不同濃度水平的混合標準溶液(質量濃度分別為1,5,10 μg/L),按1.2 進行預處理后測定,做回收試驗,每個濃度水平分別作6 次重復測定,檢測結果見表2。
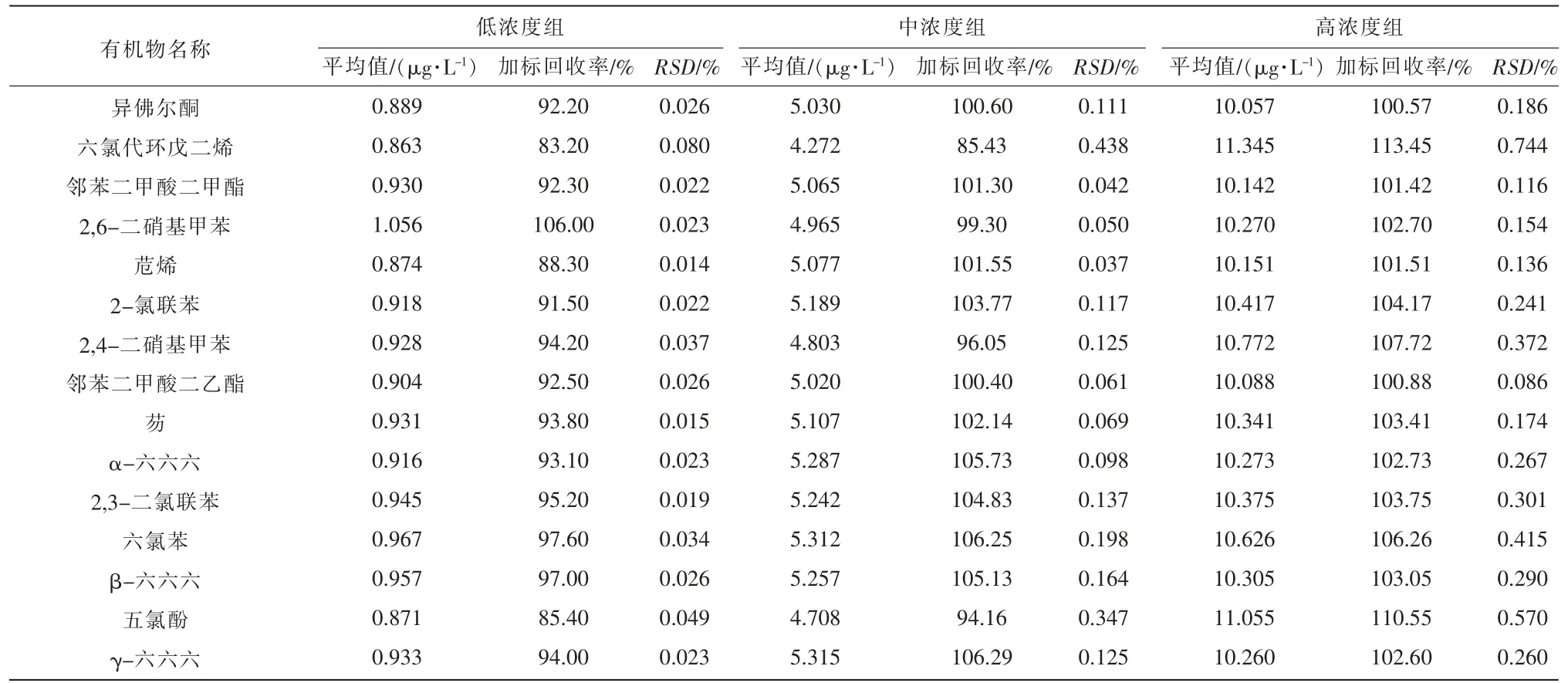
表2 49 種SVOCs 的加標回收率和相對偏差

續表
由表2 可知,49 種SVOCs 的回收率在72.50%~113.45%之間,RSD為0.010%~0.744%。其中低濃度組加標回收率范圍為72.50%~109.90%,RSD為0.010%~0.105%;中濃度組回收率范圍為85.43%~106.29%,RSD為0.042%~0.438%;高濃度組加標回收率為97.46%~113.45%,RSD為0.744%~0.086%,說明該方法對不同濃度范圍內的化合物都具有較好的回收率和精密度。
2.6 實際樣品檢測
選取南京市水源水和自來水管網末梢水為實測樣品,其中3 個為水源水樣品、7 個網末梢水樣品,按照上述實驗方法進行檢測,均未檢出半揮發性有機化合物,水質符合國家標準要求。
3 結論
本試驗采用快速固相萃取富集、凈化、濃縮水樣,結合GC-MS 聯用技術,建立了同時測定飲用水中49種SVOCs 的方法,與GB/T 5750—2006 方法比較,具有有機溶劑用量小、靈敏度高、富集率高、操作簡單、多組分同時定性定量測定等優點,而且檢出限、線性關系、回收率和重復性等方法學指標均能滿足水中痕量SVOCs 的檢測要求,可為水質中痕量半揮發污染物的快速監測與治理提供技術支持。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
