ATP1A3基因突變致兒童快發病性肌張力障礙-帕金森綜合征一例并文獻復習
2021-07-06 09:36:56康慶云廖彩時廖紅梅陳波楊理明
中國現代神經疾病雜志 2021年4期
康慶云 廖彩時 廖紅梅 陳波 楊理明
ATP1A3基因突變所致快發病性肌張力障礙-帕金森綜合征(RDP)[1]是一種肌張力障礙疊加綜合征,屬國際肌張力障礙分類12型,其臨床特征鮮明,以數小時至30天快速發病的肌張力障礙和帕金森綜合征為特征[2]。ATP1A3基因突變所致RDP是一種臨床極為罕見的運動障礙性疾病,目前世界范圍內報道的病例僅61例,國內至今僅3篇文獻共6例患者被報道[3-5]。本文回顧分析1例兒童期發病、經基因檢測確診的ATP1A3基因突變所致RDP患兒的臨床資料,通過復習相關文獻總結該綜合征特點,以期提高臨床醫師對該病的認識水平。
病例資料
患兒 女性,4歲9個月。因發熱4天伴乏力、意識障礙3天,于2019年3月6日入院。患兒4天前因“受涼”而誘發發熱、頭痛及咽痛,次日晨起出現肢體乏力、豎頭不穩、不能直立、嗜睡等癥狀與體征,遂至當地醫院就診,腰椎穿刺腦脊液及頭部MRI檢查均未發現明顯異常,考慮“腦干腦炎”,予抗病毒中藥炎琥寧(80 mg/d)和多種維生素對癥支持治療,連續治療3天癥狀無緩解且逐漸出現吞咽困難、失語等癥狀,為求進一步診斷與治療至我院就診,以“顱內感染”原因待查收入院。自發病以來呈嗜睡狀態,大小便正常,體重無明顯變化。
既往史、個人史及家族史 系孕2產2、足月順產,智力、運動發育里程碑正常,3月齡抬頭、7月齡獨坐、12月齡獨走。既往體格健康,發病前可自行進食、如廁,與他人交流無語言障礙。父母體格健康,無同類疾病或家族遺傳性疾病病史。
入院后體格檢查 患兒體溫為36.5℃,心率為102次/min,呼吸為21次/min,血壓為130/80 mm Hg(1 mm Hg=0.133 kPa);一般內科檢查無異常。神經系統檢查呈嗜睡狀態,哭鬧不安,無言語交流;頸軟;眼瞼無下垂、無眼震,眼球活動正常;口角無歪斜,咽反射減弱;豎頭不穩、獨坐不能,粗測雙上肢肌力2級、雙下肢肌力3級,四肢肌張力低下;雙側膝反射正常,跟腱反射正常,Kernig征、Brudzinski征均呈陰性,左側Babinski征陽性、右側為陰性。
輔助檢查 實驗室檢查血尿便常規、肝腎功能、心肌酶譜、電解質及血糖等項指標均于正常值范圍;乳酸、銅藍蛋白、血尿串聯質譜分析、腦脊液檢查無異常;血、腦脊液自身免疫性腦炎抗體呈陰性。腦電圖背景節律慢化,肌電圖無異常。頭部MRI(平掃和增強)、fMRI、脊髓MRI(平掃和增強),以及胸部X線檢查均無異常所見,眼底、腹部彩超檢查正常。入院15天(2019年3月21日)行圖片詞匯測試,原始總評分為10,百分位數<1,提示存在認識功能減退可能。考慮患兒肌張力障礙累及部位有明顯的頭腿梯度差(面部>上肢>下肢),為明確診斷,經患兒父母知情同意于入院22天(2019年3月28日)采集患兒及其父母外周靜脈血2 ml,采用外顯子芯片捕獲+高通量測序技術進行第二代基因組DNA全外顯子組測序(WES,北京金準基因科技有限責任公司),根據測序結果設計聚合酶鏈反應(PCR)擴增引物,采用Sanger測序并驗證。結果顯示,患兒存在ATP1A3基因c.2267G>A(p.R756H)位點雜合錯義突變,家系驗證其父母均未攜帶同型雜合突變,為新生突變(圖1)。最終確診為RDP。
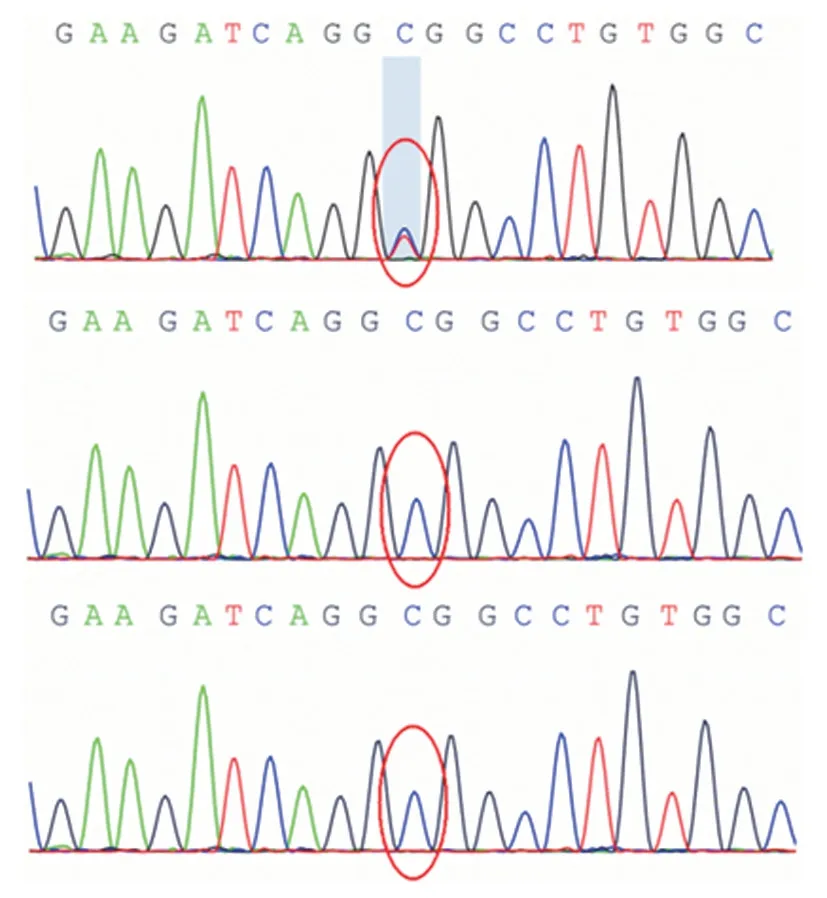
圖1 基因檢測結果 1a 先證者存在ATP1A3基因c.2267G>A(p.R756H)位點雜合錯義突變(紅圈所示) 1b,1c 先證者父母不存在ATP1A3基因c.2267G>A(p.R756H)位點突變(紅圈所示)Figure1 Genetictestingfindings Theprobandhad heterozygositymissensemutationinthec.2267G>A(p.R756H)siteofATP1A3 gene(red circleindicates,Panel1a).Theproband'sfatherandmotherhadno mutationatthissite(redcirclesindicate;Panel1b,1c).
患兒入院以后,經體格檢查和各項輔助檢查考慮為腦干腦炎或吉蘭-巴雷綜合征(GBS)譜系疾病,治療原則以抗炎、調節免疫為主。予以靜脈注射免疫球蛋白400 mg/kg(1次/d×5 d)和奧拉西坦1 g/d(1次/d×10 d)靜脈滴注,同時輔助神經營養藥、康復綜合訓練,連續治療12天(2019年3月18日)患兒神志逐漸清醒,可自主少量進食但流涎癥狀明顯,仍無明確的語言交流,肢體肌力有所改善(雙上肢肌力3級、雙下肢肌力4級)但豎頭不穩、獨坐不能如前;入院19天(2019年3月25日)時出現不自主甩頭動作,同時伴有肢體及軀干舞蹈徐動樣動作,此時肢體肌力雖已明顯好轉,但豎頭及獨坐不穩癥狀仍未緩解,建議予以氯硝西泮治療,但未獲得患兒父母同意。2019年5月6日經基因檢測確診為RDP,開始規律服用氯硝西泮0.10mg/kg(2次/d),連續治療26天(2019年6月1日)后可獨坐;72天(2019年7月17日)可獨自行走數步,言語交流呈單詞式且吐詞不清;2.50個月(2019年7月20日)可獨自行走但呈寬基底步態;4個月(2019年9月4日)能自主持勺進食;5個月(2019年10月15日)可雙足跳,自主穿、脫衣但協調性稍差,面部表情欠豐富,可說短句,發音略含糊。末次隨訪時(2019年11月30日)肢體肌力基本如常,但協調性稍差,語言表達欠流暢,構音障礙。
討 論
筆 者 以“RDP”、“Rapid-onsetdystonia parkinsonism”、“DYT-12”、“快發病性肌張力障礙-帕金森綜合征”等詞組作為關鍵詞,分別檢索美國國立醫學圖書館生物醫學文獻數據庫(PubMed)、萬方數據知識服務平臺、中國知網中國知識基礎設施工程(CNKI)等數據庫(建庫至2019年12月),共獲得RDP相關文獻126篇61例病例,國外報道55例、國內報道6例(表1)。

表1 文獻報道的61例RDP患者臨床資料Table 1. Clinical data of 61 patients with RDP reported in the literature
RDP由Dobyns等[6]于1993年首次報告,以肌張力障礙為主要表現,多于青少年期發病,感冒、妊娠、酗酒、情感打擊等為常見誘發原因[7];典型癥狀表現為突發性肢體肌張力障礙伴隨運動遲緩、姿勢不穩、吞咽困難,以及進行性構音障礙等[8]。診斷標準包括[9]:(1)數分鐘至30天內突發快速進展的肌張力障礙和帕金森樣癥狀。(2)肌張力障礙累及部位且具有明顯的頭腿梯度差(面部>上肢>下肢)。(3)呈明顯的球部受累表現。(4)對左旋多巴治療不敏感。(5)符合常染色體顯性遺傳家族史或新生突變。本文患兒以“感染”為誘發因素,伴隨急性快速進展的肌張力障礙,受累部位具有明顯的頭腿梯度差,病程中表現為吞咽困難、失語等延髓受累癥狀,并逐漸出現舞蹈樣動作、姿勢不穩等帕金森樣癥狀,上述癥狀于發病1個月后趨于穩定,符合RDP診斷。幻覺、抑郁、焦慮等精神癥狀和認知功能減退等非運動癥狀在RDP患者中并不少見[10-11],本文患兒圖片詞匯測試結果提示存在認識功能減退。
ATP1A3基因于2000年被Pittock等[1]確定為RDP的致病基因,deCarvalhoAguiar等[12]于2004年首次克隆ATP1A3基因,可編碼鈉-鉀ATP泵上的α3亞基,在小腦、基底節、海馬、丘腦等區域神經元中呈高表達,具有維持細胞內外鈉、鉀離子交換、保證神經元電興奮性和神經遞質跨膜轉運等重要生理功能[6,13]。目前已報道的ATP1A3基因突變類型共有17種,以T613M突變位點最為常見[2,10],其次是E277K和R756H突變位點[2,14]。本文患兒存在ATP1A3基因c.2267G>A(p.R756H)位點雜合錯義突變,為RDP致病性突變,支持RDP診斷。ATP1A3基因存在外顯率不全現象[15],故部分家系病例癥狀不典型,甚至有的基因突變患者完全無癥狀,某些基因位點突變可引起兒童交替性偏癱(AHC)和(或)RDP中間型,這在D583Y、G867D、E951K、D801N及R756C位點突變中均有報道[16-18]。
除RDP外,ATP1A3基因突變與多種中樞神經系統疾病有關,如交替性偏癱、小腦共濟失調、腱反射消失、高足弓、視神經萎縮、感覺神經性耳聾綜合征、嬰兒早期癲性腦病和復發性腦病伴小腦共濟失調等[9,19-22]。不同神經系統疾病ATP1A3基因突變的位點有所不同,交替性偏癱最為常見的3種基因突變位點分別為E815K、D801N以及G947R[23],感覺神經性耳聾綜合征目前僅有E818K一種突變位點[22,24-25],而復發性腦病伴小腦共濟失調則均為第756位精氨酸變異[26]。同一基因位點突變亦可引起不同的臨床表型,D923N和E277K突變在RDP和交替性偏癱患者中均有報道[27];RDP及復發性腦病伴小腦共濟失調均存在R756H突變,好發于女性病例,肌力和肌張力下降表現更為突出[21],與本文患兒臨床特征相吻合。
本文患兒存在R756H突變,RDP臨床癥狀典型,目前與該患兒具有相同突變位點的RDP病例共報道8例[3,5,14,28-29],均于兒童期發病,表現為由發熱誘發的構音困難、肌無力同時伴有帕金森樣癥狀。其中,2例嬰兒期發病者表現為RDP重疊交替性偏癱[14,28],在病程中有二次發作,考慮臨床表型為嬰兒變異型RDP;另有3例患者來自同一家系[2],均具有幼兒期感染誘發后快速發病的特點,其中先證者病程中有二次發作,其兄及母均為典型的RDP臨床表型。既往文獻報道RDP兒童期發病罕見,且罕有二次發作[20],而包括本文患兒在內的9例ATP1A3基因R756H突變患者均于兒童期發病[3,5,14,28-29],且有4例患者病程中有二次或多次發作[3,5,14,28],提示兒童期發病的RDP可能更傾向于多次發作,并常與交替性偏癱臨床表型重疊。
目前針對RDP尚無確切有效的治療方法。根據文獻報道,大部分RDP患者對多巴胺受體激動藥(普拉克索)、擬多巴胺藥(左旋多巴)、中樞抗膽堿藥、氟桂利嗪或巴氯芬等治療不敏感[7,13];部分患者大劑量苯二氮類藥物可獲得部分緩解[1-2,13];對藥物治療效果差的患者,可嘗試行腦深部電刺激術(DBS)治療[28],但亦有報道稱DBS治療RDP是無效的[30]。另外,可根據RDP患者的臨床癥狀進行包括吞咽訓練和康復綜合治療在內的對癥支持治療,對合并抑郁、焦慮等精神癥狀者給予相應藥物對癥治療,可取得一定效果。本文患兒經氯硝基安定等藥物治療,同時輔助吞咽功能訓練及康復綜合治療,癥狀明顯改善。
RDP臨床特征鮮明,急性發病,不明原因的運動障礙,伴明顯的構音障礙、吞咽障礙等延髓受累體征,且運動障礙有明確的頭腿梯度差特點而頭部MRI等各項檢查無明顯異常,此類患者應考慮RDP的可能,及時行ATP1A3基因檢測將有助于早期診斷與治療,以及優生優育。
利益沖突 無
猜你喜歡
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
英語世界(2023年6期)2023-06-30 06:29:10
保健醫苑(2022年1期)2022-08-30 08:39:40
中國生殖健康(2020年2期)2021-01-18 02:51:26
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
小學生導刊(2018年13期)2018-06-29 03:49:00
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
獸醫導刊(2016年6期)2016-05-17 03:50:35
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
